基于SMRT 技术的水杉全长转录组分析及基因功能注释
2022-03-08刘小红
刘小红
(1.西华师范大学生命科学学院,四川 南充 637002;2.西南野生动植物资源保护教育部重点实验室,四川 南充637002)
水杉(Metasequoia glyptostroboides) 属孑遗植物,为杉科(Taxodiaceae)、水杉属(Metasequoia) 的单一种, 在植物界有“活化石”之称[1]。据史料记载,水杉起源于中生代的白垩纪,在新生代的第三纪时遍布北半球,但在第四纪的冰川期后全世界仅有极少量个体存活,直到20 世纪40 年代才在中国湖北省利川市的小河境内被首次发现[2]。水杉生长历史悠久,对于研究古生物、古气候、古地质以及裸子植物的系统演化等都具有重要意义[3]。目前,水杉已被列入国家一级保护植物名录。
水杉虽然具有重要的生物学作用,但关于它的研究主要集中在栽培引种[4-5]、生理生化[6-7]、系统发育[8]等方面,而在分子遗传水平上的研究较少[9]。转录组测序可以获得大量的基因信息[10-13],能揭示潜在的代谢途径和遗传机制,可为其进一步的分子生物学研究提供依据。
关于转录组测序,以Illumina/Solexa、Roche/454和ABI/SOLiD 为代表的二代测序平台具有测序时间短、成本低、准确性高、高通量等优点,目前在差异基因表达方面被广泛应用[14-15]。但二代测序读长较短,在没有参考基因组的情况下难以获得基因的全长序列信息。而近几年发展起来的三代测序技术已经成为更好的选择,如PacBio 测序平台的单分子测序SMRT(single-molecule real-time)技术已经成为获得全长转录序列的首要选择。三代测序技术的主要优点是读长较长(平均读长可达20 kb), 对于逆转录生成的全长转录本不需要对其进行片段化处理,可以直接作单分子测序获得全长序列信息[16-17]。全长转录组测序技术已被用于许多植物的全长转录组分析[18-20],但在水杉植物中还少见相关报道。基于此,本研究拟以来自湖北省利川市的原生水杉种为材料,采用SMRT 三代测序技术对全长转录本进行测序分析,以获得相应的遗传信息,旨在为后续基因克隆及基因功能鉴定提供依据。
1 材料与方法
1.1 材料
选取来自湖北省利川市林业科学研究所(108°96′E,30°28′N) 的原生水杉种为材料,在植株长至10 cm 左右时,分别取其根、茎、叶,液氮速冻保存。
1.2 方法
1.2.1 总RNA 的提取及其质量检测
水杉根、茎、叶的总RNA 的提取都用RNeasy Plus Mini Kit(Qiagen, Valencia, CA, USA) 完成。对提取得到的总RNA 先用Nanodrop 2000 和Agilent 2100 分析其浓度,测定OD260/280、OD260/230、25S/18S和RIN 值,再用1.0%的琼脂糖凝胶电泳分析是否有DNA 污染和RNA 降解现象。
1.2.2 SMRT 测序
将检测合格的水杉根、茎和叶的总RNA 等量混匀,经mRNA 纯化、反转录及SMRT 测序后得到全长转录组原始数据(raw data)。该工作由北京诺禾致源科技股份有限公司的PacBio 测序平台完成。
1.2.3 raw data 及subreads 统计
原始数据经质量控制处理后进行subreads 的统计分析,包括有效插入片段subreads 数据量大小、有效插入片段subreads 条数、平均subreads 长度及N50(将得到的subreads 按照长度从大到小排序,依次累加subreads 的长度,直至其长度不小于总subreads 长度的50%)的长度。绘制subreads 长度分布图。
1.2.4 全长转录本分析
选用SMRT Link 5.1 对全长转录本进行统计,按默认参数设置,统计以下项目:环形一致性序列(CCS) 数量;全长非嵌合reads 数量;全长非嵌合reads 平均长度;聚类分析之后得到的一致性序列的reads 数量。
1.2.5 转录本去冗余
利用CD-HIT[21]通过序列比对聚类法去除冗余,输出1 个非冗余的序列文件,去冗余后的基因即为unigenes。对转录本去冗余前后长度频数分布情况和含有相同转录本拷贝数的基因数量进行统计。
1.2.6 基因的功能注释
为了得到全面的基因功能信息,对去冗余之后的基因进行数据库(包括NR、NT、Pfam、KOG、Swiss-Prot、KEGG 以及GO)功能注释,分析这些基因在数据库中的注释情况,并利用NR 数据库绘制物种分布图和基因功能的GO 分类统计图。
1.2.7 基因的结构分析
利用Angel[22]进行CDS(coding sequence) 预测分析。该软件具有无错和容错2 种预测模式。本研究中,选用默认的容错模式,对CDS 序列的长度及其分布进行预测,绘制序列长度分布图。运用iTAK[23]对去冗余的unigenes 进行植物转录因子预测,并将注释到转录本数量最多的转录因子家族进行柱形图展示。
2 结果与分析
2.1 总RNA 的分离纯化
水杉总RNA 分离纯化后,用Nanodrop 2000和Agilent 2100 检测的结果显示:在最后加入溶剂都为30.00 μL 的条件下,叶的总RNA 浓度最高,达301.70 ng/μL,其后依次为茎的(268.80 ng/μL) 和根的(189.70 ng/μL),三者的总量分别为9.05、8.06、5.69 μg。根、茎和叶的总RNA 的OD260/280值分别为2.04、2.05、2.11,OD260/230值分别为1.25、1.10、1.47,25S/18S 值分别为1.50、1.50 和1.40,RIN 值分别为8.50、8.70、7.70。琼脂糖凝胶电泳结果显示,每个样品都有2 条明亮的条带,没有拖尾现象,表明提取的RNA 质量较好,没有DNA 污染和RNA降解现象。上述检测结果表明,提取的RNA 样品能满足建库测序对质量的要求。
2.2 raw data 及subreads 结果分析
将提取的水杉根、茎、叶3 个部位的总RNA 样品等量混匀,经mRNA 纯化、反转录及SMRT 测序,对原始数据进行质量控制处理后共得5 914 711 个subreads 和14.0 Gb subread base,平均subreads 长度为2368 nt,N50 为2569 nt。subreads 的长度分布如图1 所示。在长度为100~10 000 nt 的范围内,2300 nt附近的subreads 最多,而超过7000 nt 的则非常少。
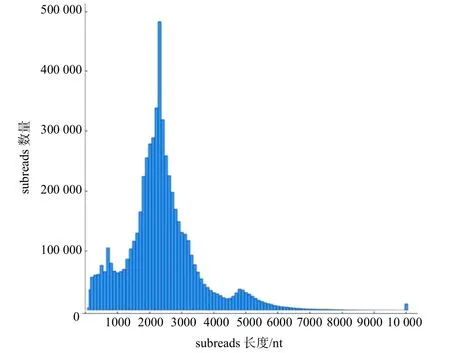
图1 水杉全长转录组subreads 的长度分布Fig.1 The length distribution of the s ubreads from the full-length transfriptome of Metasequoia glyptostroboides
2.3 全长转录本数据统计分析
采用SMRT 技术测序后共获得339 296 个CCS序列,其中用于后续试验分析的全长非嵌合reads共计236 130 个,平均长度为2598 nt,对其作一致性序列处理,最后得到一致性reads 97 626 个。
2.4 转录本的去冗余结果分析
对97 626个一致性reads进一步作去冗余处理,去冗余前后的结果如表1 所示。小于500 bp 的转录本和基因数量较少,分别仅有739、363 个;在1~3 kb 的转录本和基因数量较多,约占总量的96.92%。去冗余后最终得到的基因数(unigenes 的数量) 为61 057 个,占总转录本的62.54%。
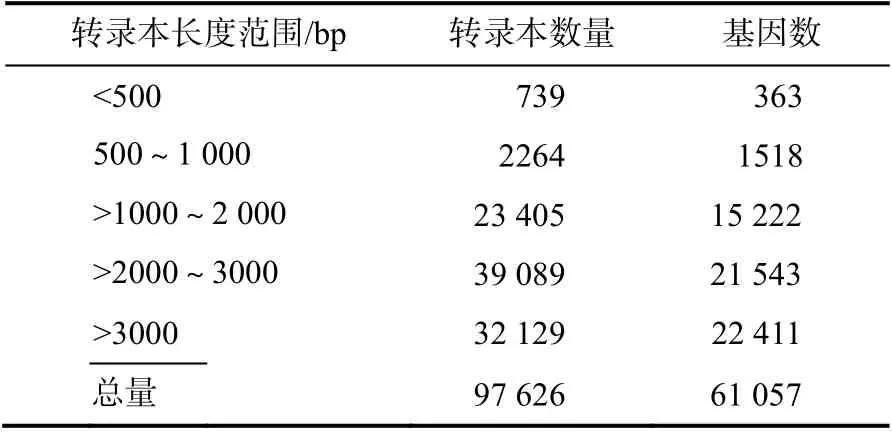
表1 水杉转录本去冗余前后的长度分布Table 1 Length distribution of the transcripts before and af ter redundancy removal in Metasequoia glyptostroboides
对去冗余的61 057 个基因作转录本拷贝数分析,结果拷贝数最少的为1 个拷贝,最多的为10个拷贝,其中1 个拷贝的基因数达51 523 个,占总量的84.39%;含9 个拷贝的基因数最少,为134个,仅占总量的0.22%。
2.5 基因功能注释结果分析
2.5.1 注释基因数量
利用NR、Swiss-Prot、KEGG、KOG、GO、NT 和Pfam 7 个数据库对水杉全长unigenes 进行功能注释,共有54 099 个基因被成功注释,被注释的基因在7 个数据库中的数量依次为52 714、42 752、51 073、32 559、24 045、31 455、24 045 个,在7个数据库中均被注释的有12 043 个。选取常用的NR、KEGG、KOG、GO 和NT 5 个数据库的注释情况绘制韦恩图(图2)。从图2 可知,在NR 数据库中注释的基因数最多,其次为KEGG 数据库,最少的为GO 数据库,在5 个数据库中均有注释的基因数为12 209 个。
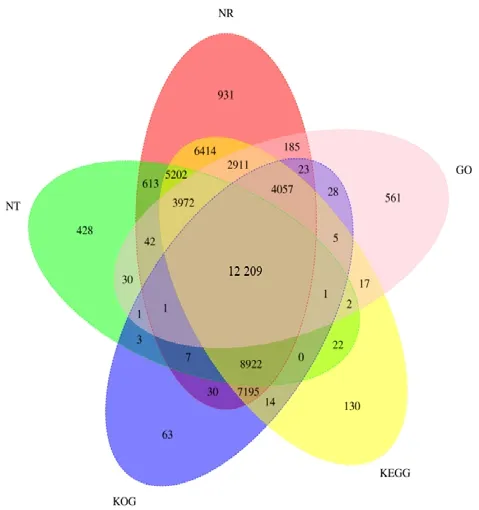
图2 水杉基因功能注释韦恩图Fig.2 The Venn diagram of gene function annotation in Metasequoia glyptostroboides
2.5.2 NR 数据库注释
通过与NR 数据库进行比对注释,共有52 714个基因被注释到568 个物种中。匹配基因数最多的是北美云杉(Picea sitchensis),达12 519 个;其次为无油樟(Amborella trichopoda) ,5888 个;排名第三的为莲(Nelumbo nucifera),3417 个;最少的为小油桐(Jatropha curcas),仅有442 个。
2.5.3 GO 分类
对上述unigenes 进行GO 注释,将注释成功的基因按照GO 3 个大类的下一级进行分类,结果显示:与细胞组分(cellular component)有关的基因有21 966 个,归属于18 个下一级分类,平均每级分类为1220 个。与生物过程(biological process)有关的基因数为42 898 个,归属于25 个下一级分类,平均每级为1716 个。与分子功能(molecular function)相关的基因有28 015 个,归属于11 个下一级分类,平均每级为2547 个。
2.6 基因结构分析
2.6.1 CDS 预测
CDS 预测结果如图3 所示。共有61 259 个CDS序列,其长度不一,最长的达6 477 nt,最短的仅为144 nt,平均长度约为679 nt。总的趋势是转录本序列越长,则转录本数量越少。
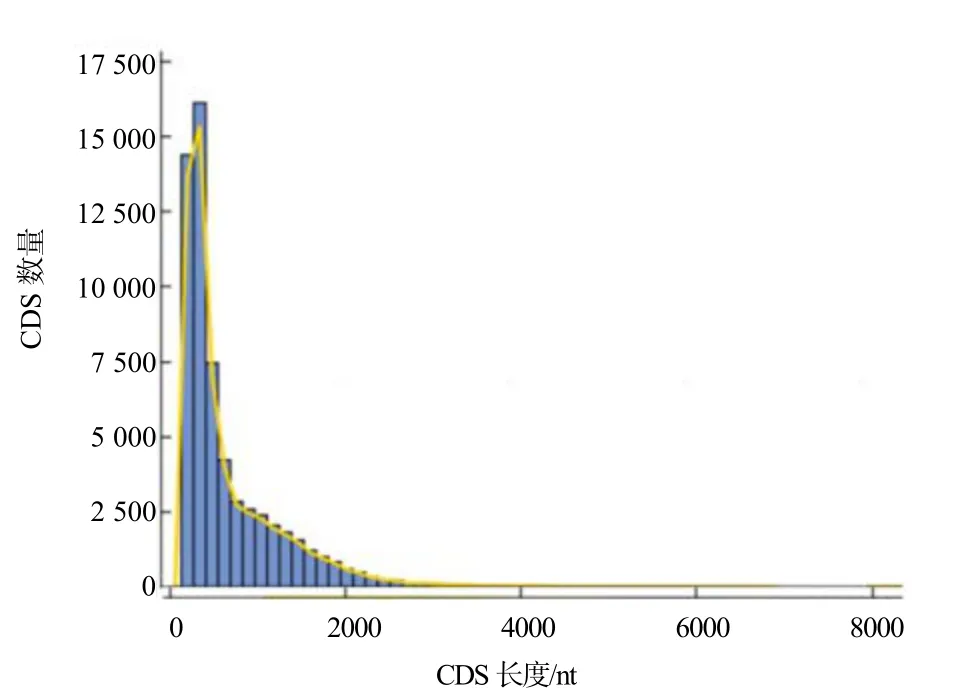
图3 水杉基因的CDS 长度分布情况Fig. 3 CDS length distribution of the genes in Metasequoia glyptostroboides
2.6.2 转录因子分析
在61 057 个unigenes 中,预测到共有2386 个表达产物为转录因子,这些转录因子可以分成29个家族,其含有的转录因子成员数最多的为C3H 家族,有234 个;其次为GRAS 家族,有194 个;最少的为HSF 家族,仅有28 个。
3 结论与讨论
全长转录组测序技术可有效获取物种转录组信息且可信度高,目前该技术被广泛应用于许多物种[24-25],有助于了解没有参考基因组的非模式生物的遗传信息[26]。本研究中,基于PacBio 平台的SMRT 测序技术被用于分析水杉幼苗全株的全长转录组测序,共获得了5 914 711个subreads和14.0 Gb的数据量。经过数据优化和去冗余处理,最后获得61 057 个基因的全长cDNA 序列信息。本研究结果与在野草莓(Fragaria vesca)[27]、紫苜蓿(Medicago sativa)[28]和高山杜鹃(Rhododendron lapponicum)[29]等中的研究结果类似。本研究中,转录本的平均长度达2598 nt,远高于二代测序的平均读长[30-31]。本研究结果还表明,不同的基因在转录组测序分析中转录本拷贝数可能不一样,其中具有单拷贝转录本的最多,占全部转录本的84.39%,总的趋势是拷贝数越多,对应的基因数量越少。
水杉虽然不是模式植物,但可以基于已有参考植物的数据库信息对本研究获得的61 057 个全长基因进行功能注释,结果共有54 099 个基因被注释到7 个数据库中,被注释的基因占88.60%,其中有12 043个同时被7个数据库注释,占总量的19.72%。通过NR 数据库的注释,发现水杉和北美云杉之间的亲缘关系较近。此外,通过GO 数据库对基因功能进行分类统计,并将这些基因分成了生物过程、细胞组分及分子功能3 大类。在基因结构分析中,CDS 预测结果显示,转录本序列越长,则其在细胞中的拷贝数越少,这与前人[20,24]在其他物种作全长转录组分析的结果一致。转录因子分析发现,获得的部分基因的表达产物其实是与细胞中基因转录调控密切相关的转录因子,与细胞中很多基因能否转录及转录强度密切相关。该结果为分析水杉基因的功能提供了分子基础。一小部分基因不能被注释,可能是因为这些unigenes 是一些新基因,在数据库中还没找到与此相似的转录本序列,有待于对这些全长转录本对应的基因的结构和功能作进一步研究。
