反刍动物瘤胃噬菌体的宏基因组学研究方法及进展
2022-03-08吴祎程周传社谭支良
吴祎程,冉 涛,周传社*,谭支良
(1.中国科学院亚热带农业生态研究所 亚热带农业生态过程重点实验室 畜禽养殖污染控制与资源化技术国家工程实验室 湖南省动物营养与生理代谢实验室 农业部中南动物营养与饲料科学观测站,长沙 410125; 2. 中国科学院大学,北京 100049;3.兰州大学草地农业科技学院,兰州 730020)
病毒(virus)广泛存在于地球各种环境中,是地球上分布最广、数量最为巨大的生命实体[1]。研究发现,大部分病毒能特异性侵染细菌,这些病毒被称作噬菌体(bacteriophage或phage)[2-3]。在噬菌体被发现后不久,研究人员便证实了反刍动物瘤胃内也存在噬菌体[4-5]。长期以来,尽管研究人员意识到病毒可能在反刍动物瘤胃微生态系统中发挥着重要作用,但受限于技术手段和瘤胃环境的复杂性,瘤胃环境中噬菌体研究进展相对缓慢[6]。目前,对瘤胃噬菌体的认知多来自于传统的培养、显微镜观察和单一噬菌体的基因组学研究[7-8],对其在瘤胃微生态系统层面的认识仍十分匮乏且片面,以致至今对其在瘤胃微生态系统中的组成和功能仍知之甚少。近年来,随着宏基因组学技术的发展,极大地推动了海洋生态环境中病毒生态学研究的发展,并取得了引人注目的成绩[9]。宏基因组学也被逐步运用于胃肠道病毒组(virome)的研究,同时刷新了研究人员对胃肠道噬菌体的复杂性和多样性的认知[10]。Manrique等[11]首次利用病毒宏基因组学技术发现肠道中81%~93%的噬菌体为新型噬菌体,由于病毒缺乏合适的标记基因和分析基准,它们无法被归类或找到对应宿主。瘤胃微生态环境复杂,瘤胃作为一个巨大的基因资源库,具有巨大的新病毒发掘空间,病毒宏基因组学技术与传统技术的联用在这一方面具有独特优势,进而推动瘤胃噬菌体相关研究。
本文主要聚焦反刍动物瘤胃生态系统中的噬菌体群落,并讨论瘤胃噬菌体研究方法及现状,及从其他生境中的噬菌体研究所得到的启发。本文还展望了病毒宏基因组学在瘤胃环境中的应用前景,以期为后续瘤胃噬菌体组研究提供科学参考。
1 瘤胃噬菌体概述
反刍家畜瘤胃内栖息着数以万亿计的微生物[12-15](图1),包括细菌、真菌、原虫、古生菌和病毒,其中病毒的数量(>108~109pfu·mL-1)仅次于细菌,且主要为噬菌体(bacteriophage或phage)[16]。瘤胃内的噬菌体具有不同形态,现有研究表明,反刍动物瘤胃中,有尾噬菌体目(Caudovirales)普遍存在,包括肌尾噬菌体科(Myoviridae)、长尾噬菌体科(Siphoviridae)、短尾噬菌体科(Podpviridae)占主导地位[17-18],以及最近被鉴定出来的阿克曼噬菌体科(Ackermannviridae)和赫雷尔噬菌体科(Herelleviridae)[19]。其实,早在20世纪60年代,“瘤胃噬菌体”一词就已进入人们视野,但对其在瘤胃微生态系统中的功能知之甚少,直到20世纪80年代末,研究人员才开始对其进行探索[20]。随着测序技术的发展,宏病毒组学技术日臻完善,借助该技术手段能够更好地探索噬菌体这个“暗物质”。已有研究表明,噬菌体在塑造瘤胃菌群结构、维持微生物多样性和调节宿主菌代谢等方面发挥着重要作用[21-22]。对反刍动物瘤胃噬菌体多样性的研究表明,噬菌体可能是通过裂解宿主细菌从而影响瘤胃细菌种群动态变化[7,23-24]。Anderson等[23]通过营养调控试验首次报道了饲粮变化对反刍动物瘤胃噬菌体群落的影响,并表明,噬菌体可通过其编码的辅助代谢基因(auxiliary metabolic genes, AMGs),经过裂解宿主细菌、产能、复制和微生物代谢的重新编程等一系列过程来影响瘤胃微生态系统[17,21,23],而其中宿主菌代谢与反刍动物饲料转化效率密切相关。众所周知,瘤胃微生物群体结构的稳定性和多样性对于反刍家畜的健康、营养、免疫和生存至关重要,因此,需要重视瘤胃噬菌体在整个瘤胃微生态系统中的地位、功能及作用。
2 瘤胃噬菌体研究方法
研究噬菌体的传统方法是显微镜观察和体外分离培养,但在实验室环境中培养的宿主菌数量有限;噬菌体还具有形态小、遗传进化快等特点,缺乏细菌所拥有的相对保守的系统发育标记,如16S rRNA基因;且感染不同宿主细菌的噬菌体具有高度特异性,不同噬菌体序列的重叠群非常小[11,25-26]。在瘤胃噬菌体的早期研究中,利用透射电子显微镜(transmission electron microscope, TEM)技术,研究人员发现,瘤胃液中存在着形态各异、高度多样化的噬菌体种群,并猜测其可能影响瘤胃内细菌的种群[27]。后来,随着DNA分析技术、脉冲场凝胶电泳技术(pulsed field gel electrophoresis, PFGE)和微生物分离鉴定技术的发展,研究人员证实,反刍动物瘤胃液中存在大量溶菌性噬菌体[7],并陆续分离鉴定了多种,包括以坏死梭杆菌(Fusobacteriumnecrophorum)[28]、白色瘤胃球菌(Ruminococcusalbus)[29]、溶纤维丁酸弧菌(Butyrivibriofibrisolvens)[30]和瘤胃拟杆菌(Prevotellaruminicola)[31]等作为宿主菌的噬菌体。TEM技术也随之被广泛用于新型噬菌体分离株的形态学表征[25, 32]。如今,宏基因组学测序技术和生物信息学的快速发展,使研究人员能更深入地了解病毒群落的结构和功能。宏基因组学(metagenomics)手段是以某一特定环境样本中的微生物群体基因组为研究对象,以功能基因筛选和序列测定分析为研究手段,以微生物多样性、种群结构、进化关系、功能活性、相互作用以及其与环境间的关系为研究目的的一种新的微生物研究方法[33]。而宏病毒组学(viral metagenomics)则是结合病毒自身特点,将宏基因组学方法应用于病毒学领域[34]。
瘤胃噬菌体宏基因组学研究,主要由病毒样颗粒(virus-like particles, VLPs)浓缩液制备与生物信息学分析两部分组成(图2)。具体步骤包括:1)瘤胃样本采集,经过滤、消化以及高速离心等技术,去除细菌等其他潜在宿主;2)对样品进行富集、纯化,得到病毒浓缩液;3)根据病毒核酸类型,选择合适的病毒核酸提取试剂盒或手工提取的方式获取病毒核酸,构建病毒基因组文库,并通过高通量测序技术进行宏病毒组测序;4)对测序所得原始数据进行质控,基于重叠区(overlap)将高质量测序读段(reads)拼接为重叠群(contigs);5)通过病毒序列鉴定软件在重叠群中鉴别、筛选病毒序列,从而了解该环境中病毒构成;6)对病毒基因组开放阅读框(open reading frame, ORF)进行预测,再通过注释工具将 ORF 与多个数据库(KEGG、COG、CAZy、CARD等)比对进行功能基因注释。
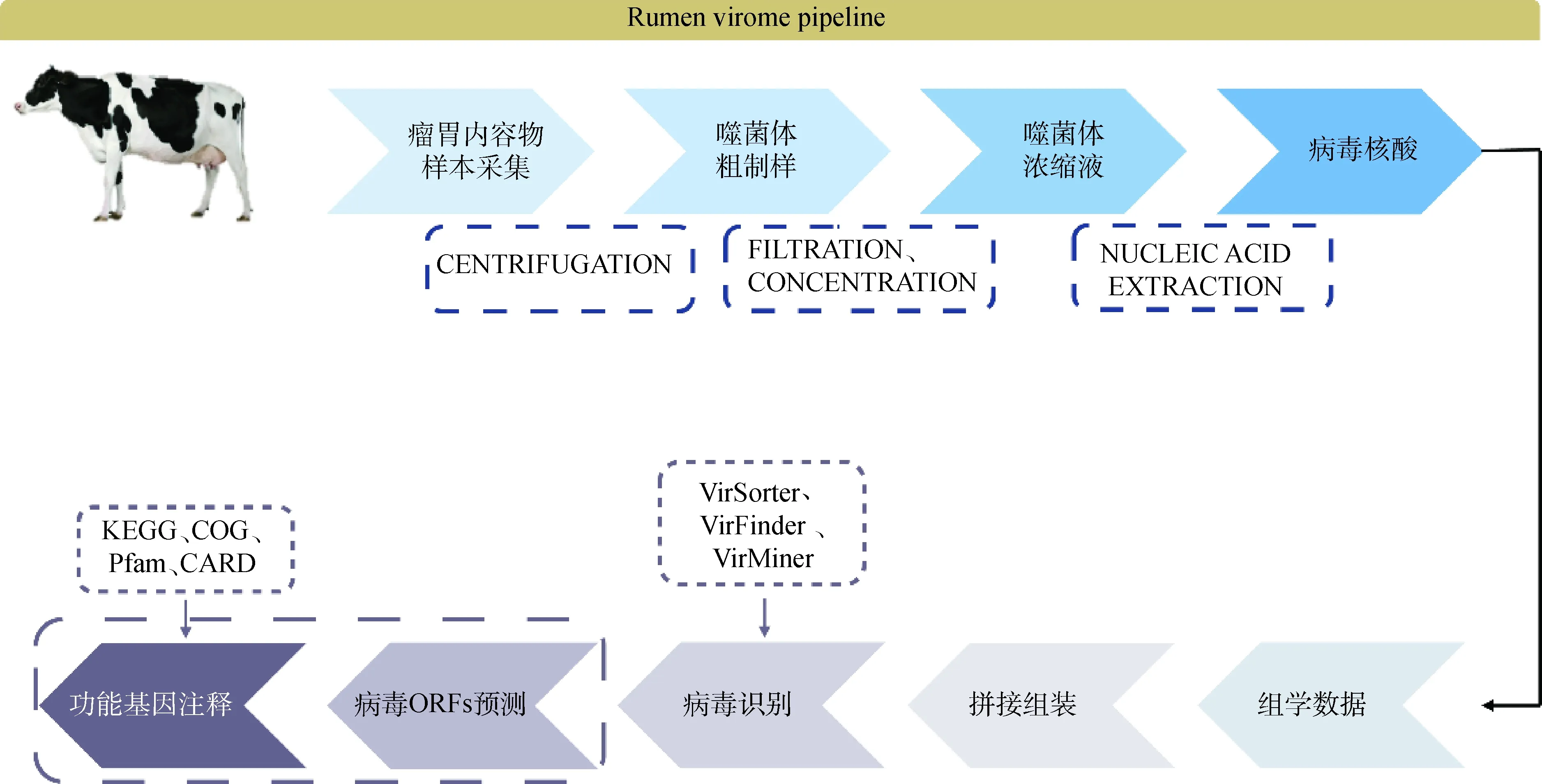
图2 瘤胃病毒组分析流程图Fig.2 Flowcharts of rumen virome analysis
2.1 样品前处理及噬菌体核酸提取
瘤胃噬菌体即为那些以瘤胃细菌为宿主菌的噬菌体,或是那些存在于反刍动物瘤胃体内的噬菌体[31]。由于采样后,细菌和噬菌体仍然彼此接触,噬菌体的侵染作用仍持续存在,长时间孵育会影响噬菌体与微生物比例[35]。因此,样品采集后要立即超低温冻存或立即对样品进行处理,将病毒样颗粒从样品中分离并纯化,用于病毒遗传物质提取。现有的提取瘤胃噬菌体核酸的方法大多参考人或小鼠肠道噬菌体及土壤噬菌体分离方法[17,23,31],截至目前,仅有一篇文献以山羊和绵羊瘤胃液作为瘤胃噬菌体来源,建立了瘤胃噬菌体富集及DNA提取方法(包括过滤、离心、沉淀及核酸提取一系列过程)[18]。
瘤胃环境异常复杂且微生物多样性高,瘤胃样品中噬菌体的分离质量直接关系到病毒宏基因组数据的质量。Hungate[36]研究表明,大约75%的瘤胃细菌紧密附着在饲料颗粒上,以细菌为宿主的噬菌体也会附着在饲料上,因此,研究瘤胃噬菌体时,需同时采集固相和液相样品,以便充分囊括瘤胃内的所有噬菌体。在从样品中分离噬菌体时,常利用噬菌体与其他微生物颗粒大小不同这一特征,借助微孔滤膜、离心等方式来实现噬菌体与其他微生物的分离。但是,实际操作中噬菌体样极易混入细菌、真菌等微生物,尤其是那些与噬菌体颗粒大小相近的微生物,使得提取的病毒宏基因组中常混有细菌、真菌等其他微生物的核酸[37]。于是,在制备噬菌体粗制样时,微孔滤膜、化学试剂等的选择对去除细菌等其他微生物的污染和保证噬菌体群体的代表性具有重要影响,进而对后续病毒核酸纯度、基因功能注释的准确性产生影响。Conceição-Neto等[38]研究发现,常见代表性病毒的大小由17到1 000 nm不等,其中,常见噬菌体的大小为125 nm左右。胃肠道病毒颗粒中绝大部分是噬菌体,为了避免细菌的污染,在研究胃肠道噬菌体组时,常选择孔径为0.22或0.45 μm的微孔滤膜,当样本环境复杂时,为避免遗漏巨型病毒信息,常选择0.45 μm的微孔滤膜,但为了更有效地防止噬菌体宿主菌污染,0.22 μm的微孔滤膜仍被广泛结合使用[39]。Friedersdorff等[30]选择0.45和0.22 μm两种孔径的微孔滤膜依次过滤瘤胃液样品,最终分离得到5种瘤胃溶菌性噬菌体,并对其进行了全基因组学测序。同样,Namonyo等[18]在建立瘤胃病毒DNA提取方法时也选用了0.45和0.22 μm两种孔径的微孔滤膜依次进行过滤。而Klieve等[31]只选择了孔径为0.45 μm的微孔滤膜对瘤胃相关样本进行过滤。虽然现有文献表明暂未鉴定出巨型瘤胃噬菌体,但并不排除技术手段造成的误差,为了更全面地研究瘤胃环境噬菌体群落,研究人员仍需根据研究目的谨慎选择微孔滤膜的孔径。
在获得含噬菌体的粗制样后,常利用聚乙二醇(polyethylene glycol,PEG)配合不同浓度的NaCl来沉淀噬菌体颗粒以实现噬菌体的浓缩。较为常用的PEG和NaCl用量为25% PEG6000(w/v)和1.0 mol·L-1NaCl[18];10% PEG8000和0.5 mol·L-1NaCl[31]及20% PEG6000和25 mol·L-1NaCl[30]。添加PEG后,常需在低温下(4 ℃)孵育过夜,以使噬菌体充分沉淀,经过高速离心(13 000×g, 30 min, 4 ℃; 12 000×g, 30 min, 4 ℃或52 350×g, 10 min, 4 ℃)后,倒掉上清液,获得的沉淀即为噬菌体样品。根据试验需求,该样品可以直接用于后续试验,也可经超速离心进一步纯化后用于后续试验。较为常用的是氯化铯(CsCl)密度梯度离心等,此法可对特定密度范围内的噬菌体进行纯化,并根据密度将VLPs分层。Carroll-Portillo等[40]的测序结果表明,用氯化铯密度梯度离心法富集的VLPs样品宿主细菌核酸去除率比未经富集的高,但氯化铯密度梯度离心法在富集样品时,对不同密度的噬菌体有明显的偏好性。Cordova等[41]也表明,某些噬菌体会由于氯化铯产生的渗透压胁迫(osmotic shock)而导致噬菌体核酸丢失,因此,研究人员会选择性采用氯化铯密度梯度离心法。
与瘤胃微生物组研究类似,病毒组研究也需先从分离得到的样本中提取遗传物质。鉴于DNA病毒在瘤胃环境中的优势地位,目前,对于瘤胃噬菌体的研究主要集中于DNA病毒的核酸提取[20]。噬菌体核酸提取主要有两种方式:第一种是试剂盒法,如Roche high pure viral nucleic acid large volume kit (Roche, Switzerland)[18]、QIAamp Ultra Sens Virus Kit (Qiagen)[23]和Fast DNATMSpin Kit for Soil (MP Biomedicals, Solon, OH, United States)[31]等,这类试剂盒可用于DNA病毒核酸的提取,试剂盒具有操作方便、提取纯度高且不需要直接接触有害化学试剂等优点;第二种是传统的苯酚-氯仿提取法,利用核酸、蛋白质等杂质在水相和有机相中溶解性不同而重新分配的性质来实现核酸提取,该方法价格低廉,但耗时长且提取浓度低,常需进一步纯化后才能达到测序要求[42]。试验中,若遇提取的噬菌体DNA浓度过低而不能满足测序要求的情况,可利用多重置换扩增(multiple displacement amplification, MDA)对核酸进行扩增,得到的DNA可用于构建基因文库或高通量测序[43],但扩增技术所得到的产物可能存在偏好性,还是建议提高原始噬菌体样品富集量以保证结果的准确性[3]。
2.2 高通量测序与病毒序列鉴定
高通量测序技术(high-throughput sequencing, HTS) 是对传统Sanger测序技术革命性的变革,可对数百万个DNA分子进行同时测序,并可深入地对一个物种的基因组和转录组进行整体分析,因此,也称其为下一代测序技术 (next generation sequencing, NGS)[44]。HTS具有通量高、速度快、成本低等优点,已广泛应用于宏病毒组学的研究中。近年来,牛津纳米孔技术(Oxford nanopore technology, ONT)作为新兴的单分子实时测序技术(single molecule real-time sequencing technology, SMRT)之一,具有快速制备文库,超长读取和实时数据采集等优势[45]。该技术的核心是蛋白质纳米孔,通过产生覆盖单个病毒颗粒内所有突变的基因组长度读数来获得病毒基因组。在此基础上,中国科学院微生物研究所王军研究员团队开发了一种新的工作流程(包括病毒颗粒富集、核酸的逆转录和扩增以及生物信息学分析),并首次使用ONT PromethlON平台对人类病毒组进行表征[46]。相较于传统的二代测序,ONT具有读长长的优点,因此,无需进行PCR扩增,便可以在更短时间内、更准确地从样品中生成数据。
高通量测序后首先要对原始数据进行质量控制和过滤,去除宿主核酸,再进行序列的拼接,并运用专业软件来鉴定病毒序列,确定该序列的生物群落来源,筛选出有用的基因信息。对噬菌体基因组进行组装时,一般挑选1 000 bp以上组装序列,再应用一系列软件对病毒contig进行鉴定。表1列出了常用于噬菌体鉴定的生物信息学分析工具。
从同时包含噬菌体及其宿主的混合基因组数据集中准确识别噬菌体,是分析样品中噬菌体组成的关键步骤。目前,有非常多基于序列或基于重叠群对病毒进行鉴定的生物信息学分析软件。VirSorter(https://github.com/simroux/VirSorter)可利用概率模型和大量病毒数据最大限度地检测新病毒,虽然该工具在很大程度上依赖对已有病毒基因组的相似性搜索,但它有一个显著优势,即可手动管理病毒参考基因组数据库,并补充了以海水、人体肠道、肺部组织及唾液样本为来源的宏病毒组序列[47]。2017年,Ren等[48]开发的VirFinder(https://github.com/jessieren/VirFinder)是通过利用细菌和病毒在K-mer上的差异,将病毒从宏基因组数据集中识别出来,与VirSorter相比,它能鉴定出更多潜在病毒,尤其是大小在1 000 bp以下的病毒contig。但VirSorter和VirFinder进行噬菌体分析的功能仍相对有限,在鉴定噬菌体重叠群后,无法预测噬菌体与宿主间的相互作用。MARVEL(https://github.com/LaboratorioBioinformatica/MARVEL)是2018年由Amgarten等[49]开发的工具,以其对Caudovirales目dsDNA病毒的有效注释能力而为人所知,但该工具鉴定RNA病毒的错报率非常高。2019年开发的VirMiner(https://github.com/TingtZHENG/VirMiner)则针对噬菌体分析功能做出了改进,可通过预测模型从宏基因组数据集中预测噬菌体重叠群并进行下游分析,包括功能基因注释及噬菌体与宿主间关系预测等[50]。VIBRANT(https://github.com/AnantharamanLab/VIBRANT),是2020年开发的利用迭代注释进行病毒识别的工具,它能高效识别以细菌为宿主的多种病毒(包括dsDNA、ssDNA、dsRNA和ssRNA病毒),并从宿主序列中提取前病毒区域,从而预测到其他分析工具无法预测到的前病毒(prophage),且能解析环境中不同病毒之间新陈代谢的能力[51]。2021年,Guo等[52]改进并发布了VirSorter2(https://github.com/jiarong/VirSorter2),但VirSorter仍是最被广泛使用的工具。现有的每种工具都有其局限性,应根据实际需求选择使用或联合使用不同的软件。
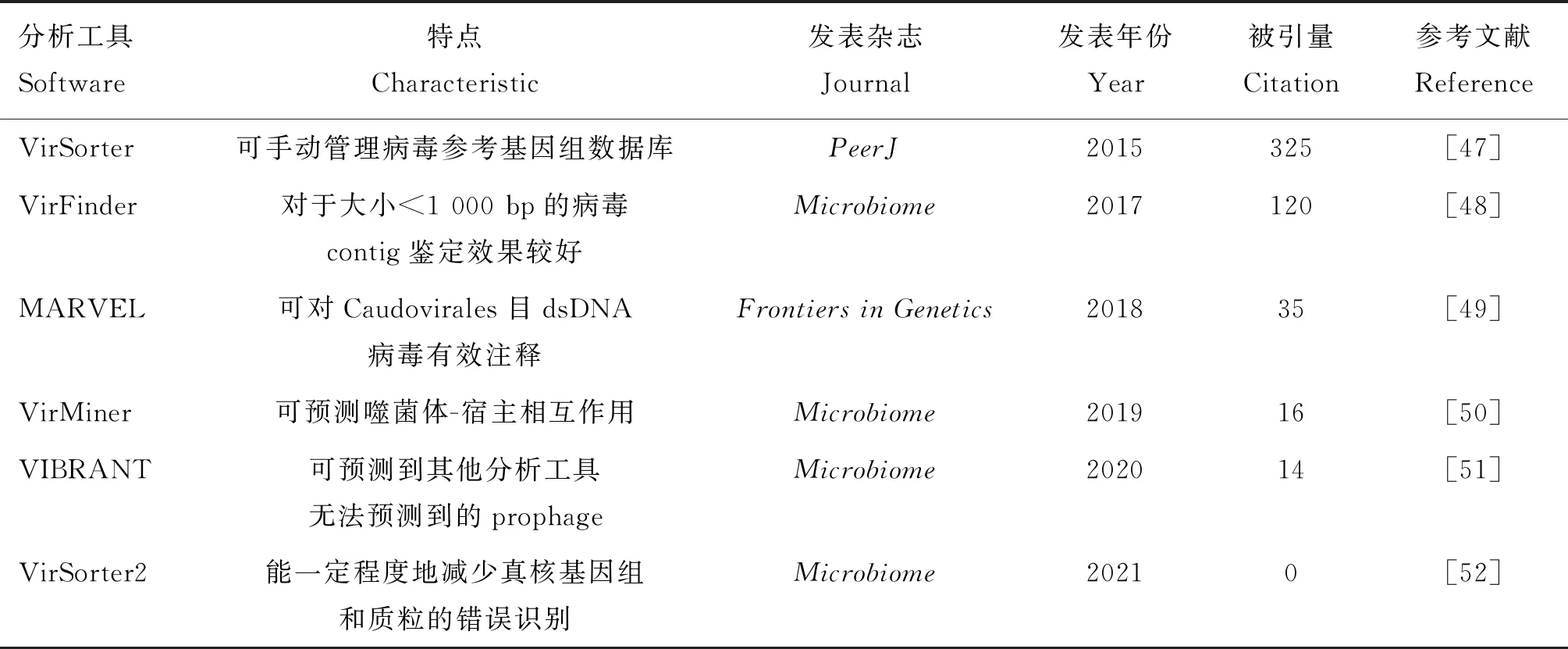
表1 病毒鉴定工具
2.3 病毒功能基因注释
使用BLAST软件进行基因预测,并将获得的非冗余基因组与RefSeq病毒数据库的参考序列进行比对,获取该基因的功能信息[53]。通过对病毒功能基因注释,能够对深入认识病毒个体生命过程提供理论基础,还有助于了解病毒群落的生态过程及与宿主群落的互作网络,从而阐释病毒与宿主间复杂的相互作用机制。
京都基因与基因组百科全书(Kyoto encyclopedia of genes and genomes,KEGG)是一个系统分析基因功能的知识库,KEGG具有强大的图形功能,可利用图形来介绍众多代谢途径(包括各种代谢通路、合成通路、膜转运、信号传递、细胞周期以及疾病相关通路等)以及各个途径间的关系,这样可以直观地反映基因与相关代谢的关系[54]。蛋白质直系同源簇(clusters of orthologous groups of proteins,COG)数据库,可将不同物种中的直系同源基因进行聚类,通过在不同物种中建立相关同源蛋白簇来预知位置蛋白质的功能,同时也为分子系统发育分析提供数据基础。2015年,Galperin等[55]基于完整噬菌体基因组中的编码蛋白系统进化关系构建了 POG (phage orthologous groups)数据库,可在双链DNA噬菌体的全基因组序列中鉴定出保守的直系同源簇。Pfam 数据库是蛋白质家族的数据库,根据多序列比对结果和隐马尔可夫模型(hidden markov model, HMMs),可查询蛋白质家族或蛋白结构域的注释、结构及多序列比对信息,被广泛用于基因功能注释[56]。抗生素的滥用对畜牧业造成了深远影响,虽然抗生素对病毒并无直接作用,但有研究发现,瘤胃噬菌体携带抗生素耐药性基因[23],因此,对病毒功能基因序列进行耐药性检测就显得尤为重要。抗生素综合研究数据库(comprehensive antibiotic resistance database,CARD) 可在细菌耐药性的分子基础上,提供参考 DNA 和蛋白质序列、检测模型和生物信息学工具,通过与该数据库进行比对,可用于关联抗生素模块及其目标、抗性机制、基因变异等信息与耐药基因相关的注释信息[57]。
3 瘤胃噬菌体基因组研究趋势及进展
借助Web of Science核心合集以“rumen bacteriophage”、“rumen phage”或“rumen virus”为关键词进行检索,2000—2021年时间段内共有108篇相关文献,且发文量呈现整体上升趋势(图3a),2021年发表的5篇文献并未在图中标明。其中,美国、澳大利亚、印度、新西兰和加拿大的发文量分别为33.66%、15.39%、9.62%、8.65%和7.69%,而我国在瘤胃噬菌体这一领域的研究严重滞后。所发表的关于瘤胃噬菌体的文章中,基于宏基因组学的文献仅有7篇[17-18,21,23-24,58-59],涉及研究动物包括水牛、奶牛、绵羊、山羊和麋鹿,且很少涉及瘤胃噬菌体群落动态变化规律及瘤胃噬菌体在整个瘤胃微生态系统中的作用。随后,利用VOSviewer进行关键词密度聚类,并进行可视化分析[60](图3b),图中结点越接近黄色,表示此为研究热点,可发现瘤胃环境病毒研究热点为噬菌体及宏基因组层面。而以“gut phage”或“gut virus”作为关键词检索,在2000—2021年时间段内共有4 449篇相关文献,这说明,瘤胃噬菌体是一个极具发掘空间的研究领域。
对病毒进行全基因组测序(complete genome sequencing),又叫单病毒基因组学(single-virus genomics, SVG),可获得特定病毒(噬菌体)的全部核酸序列。SVG即以纯培养的病毒为研究对象,进行核酸提取、文库构建并对单个病毒进行全基因组测序,SVG从头组装可以规避病毒遗传物质多样性的问题,从而构建更完整的病毒基因组,以获得更全面的病毒数据库的信息[61]。目前,仅有两篇基于SVG来研究瘤胃噬菌体的文章,表2综述了噬菌体及其宿主信息。
Friedersdorff等[30]以瘤胃拟杆菌(Prevotellaruminicola)、白色瘤胃球菌(Streptococcusbovis)及瘤胃链球菌(Ruminococcusalbus)为宿主,采用双层平板法(plaque assay)从瘤胃相关样品(废水、牛粪及瘤胃液)中分离出5株烈性噬菌体,这是首次对瘤胃环境中以特定细菌为宿主的噬菌体的基因组研究。随后对这5株噬菌体进行全基因组测序,并通过 Glimmer和Prodigal在线工具对噬菌体全基因组序列进行潜在编码序列(coding sequence, CDS)预测和功能注释。发现在φBrb01和φBrb02基因组中,CDS 可分为DNA 复制转录(DNA replication and transcription)、DNA 包装(DNA packaging)及宿主裂解(Host lysis)3个功能组。溶纤维丁酸弧菌(Butyrivibriofibrisolvens)是瘤胃内的一种优势菌,对纤维降解和蛋白质分解等有重要作用。Friedersdorff等[30]以其为宿主,从瘤胃液及粪便中分离了5株噬菌体,并进行全基因组测序和利用 Glimmer和GeneMarkS在线工具进行功能注释。结果显示,在科水平上,瘤胃噬菌体以有尾噬菌体为主,其中长尾噬菌体科占主导地位,与前人研究结果一致[17-18]。Ceridwen属噬菌体基因组中与其他噬菌体基因组同源的ORF中,其中20个与Siphoviridae科噬菌体有同源性,1个与Podoviridae科噬菌体有同源性。另外,用PHACTS软件对噬菌体生活史进行预测发现,这10株噬菌体中仅有Arawn和Idris属噬菌体基因组中存在编码整合酶的基因,表明这两株噬菌体可能为溶源性噬菌体(lysogenic phage),其他8株噬菌体为溶菌性噬菌体(lytic phage)。值得注意的一点是,从瘤胃液中分离出的Bo-Finn噬菌体与从牛粪便中分离出的Arian噬菌体基因组序列相似性可达98.6%,它们属于同种噬菌体。
4 不同生境噬菌体研究进展
自2002年美国学者Breitbart等[62]首次对海洋病毒群(virome)进行宏基因组测序,到2020年,全球范围内的研究已报道了近20万个病毒种群[63]。目前,噬菌体的研究已然成为研究不同生境内微生物群落结构和功能的焦点,如哺乳动物肠道噬菌体与机体健康和疾病发生已受到广泛关注。关于不同生境噬菌体(如人体肠道、土壤等)的研究已证实,噬菌体是导致环境中宿主细菌死亡的主要原因[64],且噬菌体可作为水平转移基因(horizontal gene trans-fer, HGT)的重要载体,与宿主菌进行遗传物质的交换,进而调节细菌的进化和多样性;值得注意的是,HGT也是导致抗生素抗性基因(antibiotic resistance genes, ARGs)扩散的重要因素[65]。鉴于瘤胃内噬菌体研究相对滞后的现状,可以适度借鉴其他环境样本中的噬菌体研究方法并用于瘤胃噬菌体研究。

图3 瘤胃噬菌体领域年发表论文数量(a)及关键词密度图谱(b)Fig.3 Number of publications(a) and keyword dendity network map(b) in the field of rumen virus (bacteriophage)
许多环境噬菌体的研究得益于微生物多组学方法联用[66-67],取得了喜人的成果。例如,Emerson等[66]利用宏基因组和宏转录组揭示了气候变化中噬菌体对复杂碳降解的潜能。此外,Brum等[67]结合蛋白质组学和宏基因组学,从海洋环境中鉴定出多样性极高的病毒衣壳蛋白。类似的,蛋白质组学这一新兴领域在瘤胃微生态系统中的研究表明,瘤胃噬菌体介导的细胞裂解会释放微生物胞内酶,包括那些参与碳水化合物分解的微生物酶,这些酶的释放会促进瘤胃内饲料的降解[59]。
除了以上能通过组学技术挖掘出的数据外,病毒宏基因组测序结果中大部分序列为未知序列,为了充分利用这些未知数据,Brum等[67]通过对预测的蛋白质序列进行全长相似性聚类,揭示了七大洋海洋样本中存在一个核心病毒基因集,这样可以从未知序列中得到另一种“已知”结果。Swain等[22]考虑到瘤胃病毒在不同个体间存在的差异,运用类似方法发现了核心瘤胃病毒基因集。此外,Allen等[68]研究海洋微生物群落时采用了一种基于噬菌体基因组标签的方法(phage genome signature-based recovery, PGSR),从宏基因组中提取噬菌体序列,并结合病毒/细菌比(virus-to-bacteria ratio,VBR)来评估病毒对细菌群落的影响,该方法有助于研究人员了解噬菌体在塑造整个海洋微生物群落结构中的作用。但目前未见此法运用于瘤胃噬菌体研究。
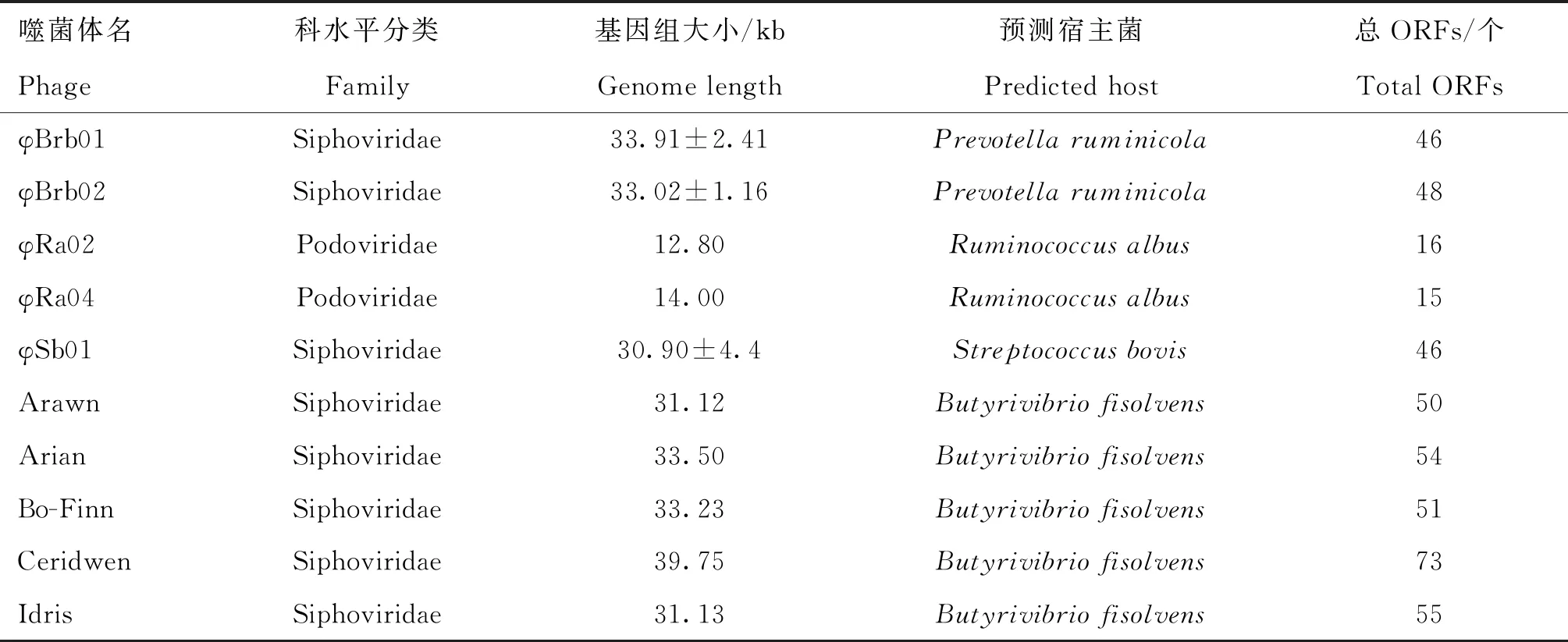
表2 已报道的获得全基因组信息的瘤胃噬菌体
值得注意的是,不同生态环境中研究方法的适用性有限。即使两个生态系统具有非常相似的病毒群落结构,他们潜在的微生态关系也不尽相同。比如,极地水体环境噬菌体和胃肠道环境噬菌体中,溶源性噬菌体较溶菌性噬菌体在数量上占有优势[69-71]。但极地海洋生态系统中,随着细菌丰度增加,溶源性噬菌体会从溶源循环(lysogenic cycle)转变为溶菌循环(lytic cycle)[69];而在肠道中,宿主菌丰度增加时,溶源性噬菌体会持续占领主导地位[70-71]。因此,在将不同生境中噬菌体研究方法及生物学概念外推到瘤胃环境时仍需谨慎。
5 瘤胃宏病毒组的前景与展望
随着宏基因组学和生物信息学的发展,研究人员可以不再需要体外分离培养噬菌体,就能获得一些分散、丰度低的病毒的结构和功能信息,也逐步增进对瘤胃内噬菌体的多样性及潜在功能的认知。但是,现阶段学术界对瘤胃噬菌体及其功能基因的科学认知仍然十分有限,笔者结合自身体会,认为今后此领域的研究应主要聚焦在以下几个方面:
5.1宏基因组学研究中,瘤胃噬菌体样品制备时主要通过微孔滤膜过滤的方式进行纯化富集,因此,难免会因为滤膜孔径选择过小而造成颗粒较大的噬菌体的遗传信息的遗漏。现阶段瘤胃噬菌体的提取、宏病毒组分析等缺乏统一技术规范,建立成熟的噬菌体分离纯化实验室标准化操作流程以及针对巨型噬菌体的优化提取和富集技术是当务之急。
5.2目前的研究主要聚焦在瘤胃DNA病毒,对于RNA病毒的研究较少。未来要利用多组学手段(如宏基因组、宏蛋白组、宏转录组和宏代谢组等)结合生物信息学技术,同步关注瘤胃RNA病毒的基因功能,尽可能获得全面的噬菌体信息,这有助于探明瘤胃整体病毒的生态功能和作用机制。
5.3鉴于噬菌体种群在个体间差异显著且噬菌体种群存在于反刍动物整个消化道,需进一步研究不同生理阶段、胃肠道不同部位噬菌体群落对微生物群落的影响。
5.4尽管病毒宏基因组学为研究人员提供了一种不需分离培养即可了解环境中全部病毒基因组信息的方法,但仍需要以瘤胃优势菌群为宿主菌,对噬菌体进行分离培养,这将有助于全面了解噬菌体的生物学特性以及它们在维持瘤胃动态平衡中的作用。
5.5关于瘤胃病毒的研究在国内处于亟待开发的阶段,瘤胃是一个复杂、多变的微生态系统,蕴藏着丰富的基因资源,有大量新病毒等待研究人员去挖掘。故需要大力发展全基因组扩增技术和测序技术,不断完善动物胃肠道病毒资源库,丰富瘤胃噬菌体基因组数据库,揭示噬菌体塑造瘤胃细菌群体结构的机制。
5.6我国于2020年开始对饲料全面禁抗,鉴于瘤胃噬菌体对瘤胃微生物的裂解作用及对瘤胃营养物质循环的影响,噬菌体制剂作为抗生素替代品将成为畜牧界的重大课题。
