综合代谢组学和转录组学分析揭示火龙果不同发育阶段的潜在颜色变化
2022-03-07杨运良苏彩虹李红义蔡岳李建勋
杨运良 苏彩虹 李红义 蔡 岳 李建勋



Integrated Metabolomic and Transcriptomic Analyses Revealing Potential Color Changes During Different Developmental Stages of Dragon Fruit
YANG Yunliang SU Caihong LI Hongyi CAI Yue LI Jianxun
Cotton Research Institute, Shanxi Agricultural University, Yuncheng, Shanxi 044000, China
Dragon fruit () development is subdivided into young (Y), transformation (C), and maturation (M) stages. In the present study, metabolomic and transcriptomic analyses were integrated to explore the molecular mechanisms underlying the physicochemical and structural changes that occur during dragon fruit development. Particular attention was paid to the factors that regulate the fruit pericarp and pulp color changes. We identified a large number of differentially expressed genes and metabolites in each comparison group. Substantial biochemical and physiological alterations were observed across the various developmental stages of dragon fruit and 214 metabolites were identified. KEGG analysis showed that flavone and flavonol signalling pathways were significantly enriched in each comparison group. The internal and external fruit color dramatically differed between the Y and C stages. The expression of Cluster-12747.5686 was upregulated, whereas those of Cluster-1339.0 and Cluster-12747.15079 were downregulated in three cytochrome P450 genes between Y and C stages. Nine differentially expressed flavonoids, including the floral / fruit pigment precursor procyanidins B1, B2, and C1, were also downregulated. This integrative analytical approach helped to explain the relationships between genotype and phenotype and clarified the numerous molecular, physicochemical, and structural changes that occur during fruit development and their relationships.
different developmental stages; dragon fruit (); metabolomic; pulp color change; transcriptomic
10.3969/j.issn.1000-2561.2022.02.001
摘 要:火龍果的发育可分为幼果期(Y)、转色期(C)和成熟期(M)。本研究综合运用代谢组学和转录组学分析方法,探讨火龙果发育过程中理化和结构变化的分子机制,特别关注调节果皮和果肉颜色变化的因素。本研究在每个对照组中发现了大量差异表达的基因和代谢物。在火龙果不同发育阶段观察到大量的生化和生理变化,鉴定出214种代谢物。KEGG分析显示,各对照组的黄酮和黄酮醇信号通路均显著丰富。Y期和C期的内外果色差异较大。在Y期、C期,3个细胞色素P450基因中,Cluster-12747.5686的表达上调,Cluster-1339.0和Cluster-12747.15079的表达下调。9个差异表达的黄酮类化合物,包括花/果实色素前体原花青素B1、B2和C1,也被下调。这一综合分析方法有助于解释基因型和表型之间的关系,并阐明了果实发育过程中发生的大量分子、理化和结构变化及其相互关系。
关键词:不同发育时期;火龙果;代谢组学;果肉颜色变化;转录组学
中圖分类号:S667.9 文献标识码:A
Dragon fruit () belongs to the Cactaceae family. It is indigenous to Latin America and the West Indies and is grown in tropical and subtropical regions. Dragon fruit is classified according to the color of its pericarp and pulp. The four main species are (red skin with white flesh), (red skin with red flesh), (red skin with super red flesh), and (yellow skin with white flesh). Of these, and are the most widely cultivated. In contrast, is produced in relatively smaller amounts. In recent years, the popularity of dragon fruit cultivation has increased among Chinese growers and consumers because of its bright color, sweet taste, high nutritional value, antioxidant content, and other features.
During its growth and development, dragon fruit undergoes complex morphological, physiological, biochemical, and genetic changes. To elucidate these alterations, researchers have conducted several studies in the early stages of dragon fruit formation. JAMALUDIN investigated dragon fruit morphophysiology, and measured structural characteristics and physicochemical indices, during development. The betacyanin content increases in maturing red-fleshed dragon fruit. HUA used transcriptomics to analyse color development in ripening red pulp dragon fruit. They identified the genes encoding betalain and proposed a preliminary betalain biosynthesis pathway in dragon fruit. WU performed metabolomic analyses on ‘Zihonglong’ dragon fruit pericarp and pulp. In the latter, the major amino acid content decreased, whereas the main sugar and organic acid content increased as fruit development progressed. Moreover, it has been reported that citramalic acid participates in betalain pigment formation in dragon fruit. HUA used proteomics to examine the white and red pulp stages of “7-1” () dragon fruit. They found that polyphenol oxidase, CYP76AD3, and oestradiol-like 4,5-DOPA dioxygenase are associated with betalain biosynthesis. Genomics, transcriptomics, metabolomics, and proteomics have continued to advance in recent years and have been applied both concurrently and separately to analyse dragon fruit. Nevertheless, the analysis of dragon fruit development via integrated transcriptomics and metabolomics remains scantly reported.
The integration of transcriptomic and metabolic profiling data may help to clarify the regulatory and metabolic events associated with plant growth and development. Several recent studies have combined multi-omics datasets and some have focused on fruit development and ripening. Here, we merged and compared analyses of dragon fruit transcriptomics and metabolomics data to identify the specific molecular regulatory responses involved in dragon fruit development.
Threeyearold dragon fruit ( ‘Mibao’) trees were used in this study. The orchard was located at Nahua Farm, Longju Town (35°01′ N, 110°89′ E, 355 m), Yanhu District, Yuncheng City, Shanxi Province, China. The spacing between trees and rows was 0.3 m×1.5 m. All trees were subjected to the same cultivation and management practices. On 30 June 2019, the flowers were artificially pollinated and marked. Fruit was sampled on day 16 (15 July 2019), 21 (20 July 2019), 26 (25 July 2019), 27 (26 July 2019), and 32 (31 July 2019) after artificial pollination (DAAP). Twenty-five tagged fruits were randomly selected and three biological replicates were taken per stage. At each sampling, the fruit was sliced cross-sectionally, frozen in liquid nitrogen, and stored at –80℃ until the subsequent analyses. Here, three developmental stages of red dragon fruit pulp (16, 27, and 32 DAAP) were designated Y, C, and M, respectively, and were used in the metabolome and transcriptome analyses (Fig. 1).
After RNA extraction, the NanoDrop device (Thermo Fisher Scientific, Waltham, MA, USA), Qubit v. 2.0 (Thermo Fisher Scientific), and Agilent 2100 device (Agilent Technologies, Foster City, CA, USA) were used to validate RNA purity, concentration, and integrity, and confirm that RNA quality was sufficient for the subsequent transcriptome sequencing. After library construction, library concentration and insert size were detected and accurately quantified with Qubit v. 2.0 and the Agilent 2100 device. Library quality was verified by qRT- PCR. High-throughput sequencing was performed with a Novaseq 6000 device (Illumina, San Diego, CA, USA). The sequencing read length was PE150.
Contents from Q30 and GC were used to evaluate the read quality using the FastQC online tool (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). GRABHERR assembled the sequence after obtaining high-quality sequencing data. The raw sequence data has been submitted to the NCBI Short Read Archive with accession number PRJNA595688. Functional annotation was aligned with the NR (RefSeq non-redundant proteins), Swiss- Prot, GO (gene ontology), KOG, and KEGG (Kyoto Encyclopaedia of Genes and Genomes) databases. FPKM (fragments per kilobase of transcript per million mapped reads) was used to calculate gene expression.
Freeze-dried samples were pulverized in a mixer mill at 45 Hz for 2 min. Subsequently, 100 mg powder from each sample was transferred to a 5 mL Eppendorf tube and extracted with 2000 µL of a methanol/water mixture (3∶1 /). The samples were vortexed for 30 s, ultrasonicated for 15 min in an ice bath, shaken at 4℃ overnight, and centrifuged at 12 000 r/min at 4℃ for 15 min. Then, 500 µL of each supernatant was dried under a gentle nitrogen flow. All residues were reconstituted in 250 µL of a methanol/water mixture (1∶1 /) with 30 s of vortexing and 15 min of ultrasonication in an ice bath. The samples were then centrifuged at 12 000 r/min at 4℃ for 15 min. The supernatants were transferred to 2 mL glass vials and stored at –80℃ until UPLC-ESI-MS/MS analysis. Samples for quality control were prepared by mixing equal aliquots of the supernatants from all samples.
Sample metabolites were qualitatively assessed according to secondary spectrum information using the in-house MetWare database and metabolite information from a public database. Metabolites were quantified by multiple reaction monitoring in triple quadrupole mass spectrometry. The triple quadrupole selected a desired fragment ion which eliminated non-target ion interference and improved quantification accuracy and repeatability. Metabolite mass spectrometry data were acquired for the various samples. The mass spectrometry peaks for the same metabolite in different samples were integrated for correction.
Characteristic ions of each substance were screened by triple quadrupole mass spectrometry. The signal intensities (counts per second) of the characteristic ions were detected and the mass spectrometry file for the sample was opened and read in MultiaQuant software. The peak area of each chromatogram peak represented the relative concentrations of the corresponding metabolites and all chromatographic peak area integral data were saved. To compare the content of each metabolite in the various samples, the mass spectrum peaks were corrected for each metabolite in the various samples to ensure the accuracy of the qualitative and quantitative analyses based on the metabolite retention time and peak type data.
Principle component analysis and partial least squares-discriminant analysis were performed to determine cultivar-specific metabolite accumulation as previously described. Variable importance in projection≥1 and fold change≥2 or≤0.5 were set to identify significantly different metabolites.
To validate RNA sequencing data accuracy, qRT-PCR was run in a model 7500 fast real-time PCR system (Applied Biosystems, Foster City, CA, USA) following the manufacturer’s instructions. The PCR amplification program was as follows: 95℃ (15 s) followed by 40 cycles at 60℃ (60 s) and 95℃ (30 s). The 2 methodwas used to calculate fold changes in gene expression. Actin was used as the reference gene for normalizing the data.
The three ‘Mibao’ Dragon fruit development stages [young (Y), transformation (C), and mature (M)] differed in terms of pulp tissue morphology and color (Fig. 1). Y-stage fruit was harvested on day 16 after artificial pollination. At that time, the pericarp was green and the pulp was white. C-stage fruit was harvested on day 27 after artificial pollination. At that time, the pericarp was dark red and the pulp was light red. M-stage fruit was harvested on day 32 after artificial pollination. At that time, the pericarp was bright red, the pulp was dark red, the overall color was darker than that of the C-stage, and the fruit was fully ripe. After observation for 6 years, it was determined that the duration of the C-stage varied with temperature and season. High temperatures in the summers can change dragon fruit color from green to red in 2 d. In contrast, similar color changes might take approximately 10 d at low temperatures in the winters.
A total of 508.8 million clean reads were acquired after quality control and data filtering. The guanine-cytosine (GC) proportion of the sequenced data was approximately 49%. More than 93% of all reads had average quality scores>30. Thus, the sequencing data were sufficiently accurate to be used in the subsequent analyses. Trinity v2.4.0 software was used to assemble the unigene. The reads were assembled into 47 855 unigenes with a mean length of 1260 bp. The median contig (N50) length was 2000 bp.
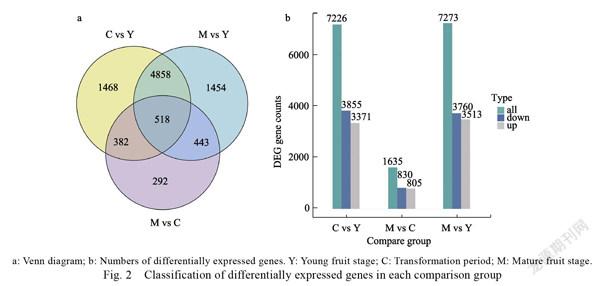
The differentially expressed genes (DEGs) involved in dragon fruit development were detected according to the criteria of <0.01 and log2 (fold change)≥1. A total of 9415 DEGs were screened across all three developmental stages (Fig. 2a). Of these, 518 were differentially expressed at each of the three developmental stages. Upregulated and downregulated genes are listed in Fig. 2b. The numbers of upregulated and downregulated genes markedly changed between the C and Y stages
(3371 upregulated vs. 3855 downregulated). There were 7273 DEGs between the M and Y stages (3513 upregulated vs. 3760 downregulated). Between the M and C stages, 1635 DEGs were detected. Of these, 805 were upregulated and 830 were downregulated (Fig. 2b).
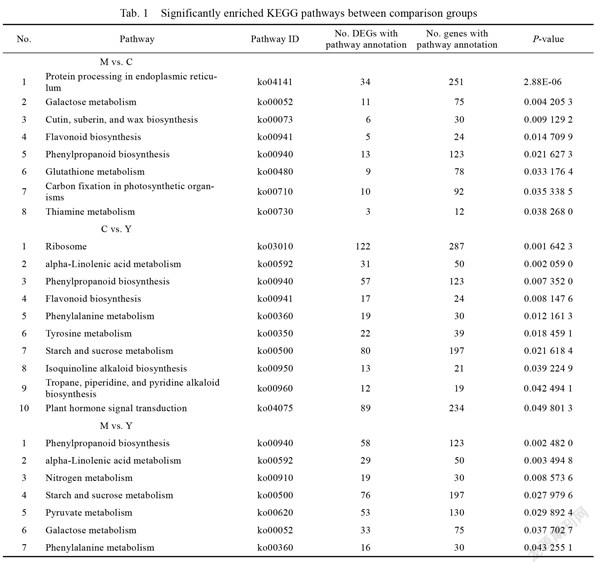
Kyoto Encyclopaedia of Genes and Genomes (KEGG) analysis revealed that phenylpropanoid biosynthesis differed significantly among the comparison groups. Protein processing in the endoplasmic reticulum, galactose metabolism, and elsewhere was significantly different in M vs. C. Ribosome, alpha-linolenic acid metabolism, and other pathways were substantially enriched in C vs. Y (Tab. 1).
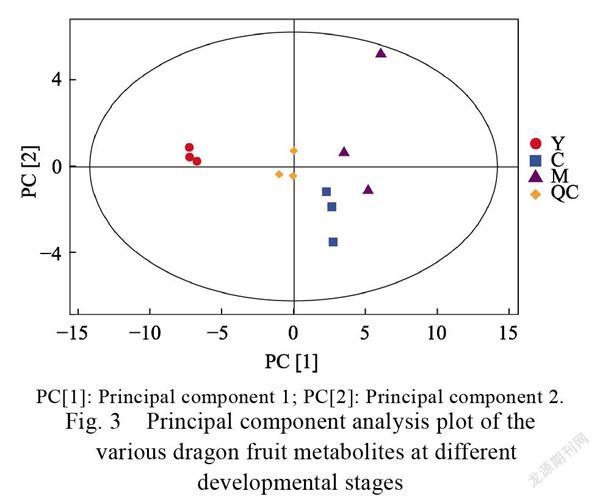
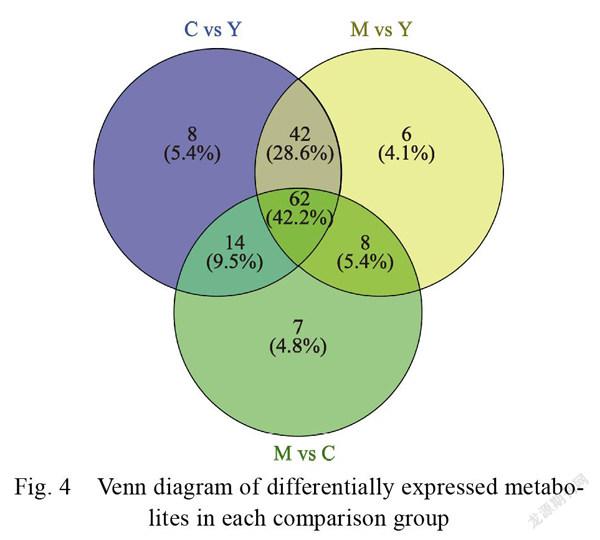
A principal component analysis of the profiles (Fig. 3) was conducted to determine the overall gas chromatography/mass spectrometry dataset structure. As expected, the three dragon fruit developmental stages differed significantly. We identified 214 metabolites in all samples. A Venn diagram was
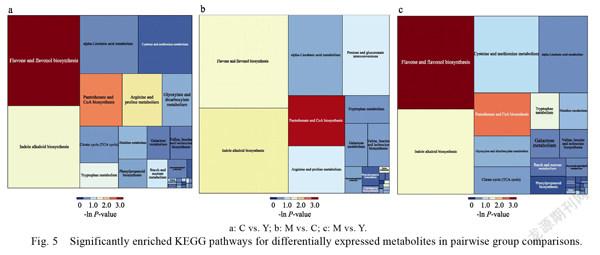
plotted to illustrate the comparisons. There were unique and shared differentially expressed metabolites (DEMs) between pairs (Fig. 4). There were 62 elements common to C vs. Y, M vs. Y, and M vs. C. A KEGG enrichment analysis was performed to identify different metabolite functions. The KEGG
results showed that flavone and flavonol biosynthesis were significantly enriched pathways in all comparison groups, especially in M vs. Y and C vs. Y (Fig. 5a, Fig. 5b, Fig. 5c).
As flavone and flavonol biosynthesis were substantially enriched signalling pathways in each comparison group, we focused on the flavonoid metabolites in dragon fruit. Nine of the metabolites were identified in the flavone and flavonol biosynthesis pathways and two were significantly enriched in M vs. Y and C vs. Y (Tab. 2).
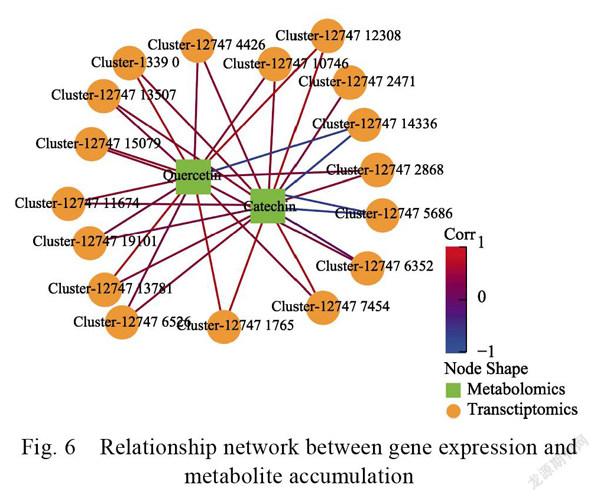
For C vs. Y, the fruit color dramatically changed and the DEGs were significantly enriched in flavonoid biosynthesis. To clarify the relationship between the metabolites and genes involved in flavonoid biosynthesis, we integrated the DEMs and DEGs, established a network, and intuitively illustrated the relationship between gene expression and metabolite accumulation (Fig. 6). Of 17 structural genes, Cluster-12747.14336 and Cluster-12747.5686 negatively correlated with catechin and quercetin, whereas the other genes showed positive correlations with the DEMs. We plotted a flavonoid biosynthesis scheme. Three cytochrome P450s (Cluster-1339.0, Cluster-12747.5686, and Cluster-12747. 15079) and three chalcone synthases (CHS) (Cluster-12747.14336, Cluster-12747.10746, and Cluster-12747.6526) were the candidate genes identified. The genes were used in the subsequent investigations. All these genes participated in dragon fruit color change. Cluster-12747.5686 (P450) and Cluster-12747.14336 (a CHS) were upregulated, whereas all other genes were downregulated.
Eleven flavonoid genes were selected and their expression levels in Y, C, and M stages were analysed by qRT-PCR to validate the RNA sequence data. qRT-PCR output for these genes was consistent with the RNA sequence results (Fig. 7) and confirmed their accuracy.
The development of high-throughput sequencing technology has led to the omics methodology, which is extensively used to elucidate biomolecular mechanisms. A study combining metabolomics and transcriptomics can provide insight into the changes in secondary metabolite profiles that occur during dragon fruit development and their associated gene expression network modifications.
In this study, the transcriptome data identified 47 855 unigenes with a mean length of 1260 bp. The median contig (N50) length was 2000 bp. Thus, we obtained higher quality data using the same technology as previous studies, which reported mean unigene lengths of 449 bp and 505 bp. Our final assembly quality was satisfactory and sufficed for the subsequent data analyses. We then conducted comparative analyses of DEGs at the various stages of dragon fruit maturation.
A total of 9415 DEGs were screened across all developmental stages (Fig. 2). Numerous DEGs were found in two comparison groups. Therefore, substantial alterations occurred across the three main developmental stages of dragon fruit. Several genes were annotated to carbohydrate-related pathways (Tab. 1). Carbohydrate metabolism plays a vital role in fruit development. More than 20 genes have been demonstrated to participate in major sugar biosynthesis in the flavedo of ripening fruit. Similar findings have been reported for grape () and peach ().
Metabolites were identified and quantified by ultra-performance liquid chromatography-electrospray tandem mass spectrometry (UPLC-ESI-MS/ MS). We screened 214 metabolites from the three dragon fruit developmental stages. KEGG analysis (Fig. 5a, Fig. 5b, Fig. 5c) showed that flavone and flavonol biosynthesis were significantly enriched in the signalling pathways of M vs. Y and C vs. Y. Flavonoids comprise anthocyanins, flavonols, flavones, flavanones, and other compounds. Anthocyanins are largely responsible for flower and fruit color. Here, the color changed dramatically between the Y and C dragon fruit stages. Nine differentially expressed flavonoid metabolites were downregulated in the C group. A network map
showed 17 structural genes, including three encoding cytochrome P450s and three encoding CHS. Four were downregulated in the C group, whereas catechin and quercetin were enriched during flavonoid biosynthesis (Fig. 5). CHS plays key roles in the biosynthesis of anthocyanins and flavonoids. (CSA024718) and (CSA029707) have remarkably different expression levels during tea flower development. CHS may account for the red coloration in crab-apple cultivars with dark red, pink, and white petals. In the present study, three CHS genes were enriched in flavonoid biosynthesis. One was upregulated and two were downregulated in the C group. However, this discovery was not altogether consistent with previous findings.
The color of red pulp dragon fruit is attributed to high betalain concentrations. A previous transcriptomic analysis indicated that the expression levels of the CytP450 genes and gradually increase during red pulp color development. These genes may regulate betalain biosynthesis. However, we found that Cluster-12747.5686 was upregulated, whereas Cluster- 1339.0 and Cluster-12747.15079 were downregulated, in the three cytochrome P450 genes from the Y to C stages. Nine differentially expressed flavonoid metabolites, including procyanidin B1, procyanidin B2, and procyanidin C1, were also downregulated.
In conclusion, this study integrated metabolomics and transcriptomics and elucidated the mechanisms by which the expression of genes, formation of metabolites, and biosynthesis of anthocyanins and flavonoids are modulated at different developmental stages of dragon fruit. Our results provide new insights into anthocyanidin and procyanidin metabolites in dragon fruit and the global transcriptional changes in the CHS and cytochrome P450 genes that regulate dragon fruit color, secondary metabolism, and ripening.
- WU Y W, XU J, HE Y Z, SHI M Y, HAN X M, LI W Y, ZHANG X W, WEN X P. Metabolic profiling of pitaya () during fruit development and maturation[J]. Molecules , 2019, 24(6): 1-16.
- DONG H S, SUNMIN L, DO Y H, YOUNG-S K, SOMI K C, SARAH L, CHOONG H L. Metabolite Profiling of red and white pitayas ( and ) for comparing betalain biosynthesis and antioxidant activity[J]. Journal of Agricultural and Food Chemistry, 2014, 62(34): 8764-8771.
- JAMALUDIN N A, DING P, HAMID A A. Physico-chemical and structural changes of red-fleshed dragon fruit () during fruit development[J]. Journal of the Science of Food and Agriculture, 2011, 91(2): 278-285.
- HUA Q Z, CHEN C J, CHEN Z, CHEN P K, MA Y W, WU J Y, ZHENG J, HU G B, ZHAO J T, QIN Y H. Transcriptomic analysis reveals key genes related to betalain biosynthesis in pulp coloration of [J]. Frontiers in Plant Science, 2015, 6(5): 1179.
- HUA Q Z, ZHOU Q J, GAN S S, WU J Y, CHEN G B, LI J Q, YE Y X, ZHAO J T, HU G B, QIN Y H. Proteomic analysis of s reveals metabolic pathway changes[J]. International Journal of Molecular Sciences, 2016, 17(10): 1606.
- HUANG H H, XU L L, TONG Z K, LIN W P, LIU Q P, SHENG L J, ZHU M Y. characterization of the Chinese fir () transcriptome and analysis of candidate genes involved in cellulose and lignin biosynthesis[J]. BMC Genomics, 2012, 13(1): 648.
- WANG Z J, CHEN J H, LIU W D, LUO Z S, WANG P K, ZHANG Y J, ZHENG R H, SHI J S. Transcriptome characteristics and six alternative expressed genes positively correlated with the phase transition of annual cambial activities in Chinese fir [ (Lamb.) Hook][J]. PLoS One, 2013, 8(8): e71562 .
- ZHANG C S, ZHANG H W, ZHAN Z X, LIU B J, CHEN Z T, LIANG Y. Transcriptome analysis of sucrose metabolism during bulb swelling and development in onion ( L.)[J]. Frontiers in Plant Science, 2016, 7: 1425.
- PATEL M, MANVAR T, APURWA S, GHOSH A, CHIKARA S K. Comparative transcriptome analysis and metabolic pathway studies of flavedo from naive stage to ripened stage[J]. Molecular Biology Reports, 2014, 41(5): 3071-3080.
- HAN Y P, VIMOLMANGKANG S, SORIA-GUERRA R E, ROSALES-MENDOZA S, ZHENG D M, LYGIN A V, KORBAN S S. Ectopic expression of apple genes contributes to anthocyanin accumulation in the mutant grown under nitrogen stress[J]. Plant Physiology, 2010, 153(2): 806-820.
- ZHANG H C, LIU J M, YU H Y, GAO S L. Enhanced flavonoid production in hairy root cultures of Fisch by combining the over-expression of chalcone isomerase gene with the elicitation treatment[J]. Plant Cell Reports, 2009, 28(8): 1205-1213.
- ROTHENBERG D O, YANG H J, CHEN M B, ZHANG W T, ZHANG L Y. Metabolome and transcriptome sequencing analysis reveals anthocyanin metabolism in pink flowers of anthocyanin-rich tea ()[J]. Molecules, 2019, 24(6): 1064.
- CHENG M N, HUANG Z J, HUA Q Z, SHAN W, KUANG J F, LU W J, QIN Y H, CHEN J Y. The WRKY transcription factor HpWRKY44 regulates CytP450-like1 expression in red pitaya fruit ()[J]. Horticulture Research, 2017, 39(4): 17039.
-
收稿日期 2021-07-05;修回日期 2021-07-26
基金项目 山西省科技厅面上青年基金项目(No. 201701D221204);山西省农业科学院特色农业技术攻关项目(No. YGG17050);
运城市科技局基础研究项目(No. YCKJ-2021041)。
作者简介 杨运良(1986—),男,硕士,助理研究员,研究方向:设施果树栽培及生理生化。*通信作者(Corresponding author):
李建勋(LI Jianxun),E-mail:lijxyc@163.com。
