水分子在药物设计中的作用
2022-03-06周洋陆小云
周洋,陆小云
(暨南大学药学院,广东 广州510632)
随着结构生物学的快速发展,大量与疾病密切相关的蛋白结构得到解析,极大地促进了基于结构的药物设计(structure-based drug design,SBDD)的发展。利用SBDD进行化合物的结构优化迭代,已经在药物研发过程中发挥不可替代的重要作用。大量的蛋白晶体结构表明,在药物结合口袋中,经常有水分子出现,这些水分子对于化合物与靶标的结合起到关键作用。然而在以往的大多数SBDD中,对于利用这些水分子指导药物设计的应用还不够广泛[1]。
近年来,通过替换口袋中的水分子,或设计化合物与水分子形成相互作用,指导化合物设计改造,已经在提高化合物亲和力、选择性以及改善其药动学性质等方面有了越来越多的应用,如Kirsten大鼠肉瘤病毒癌基因同源物(Kirsten rat sarcoma viral oncogene homolog,KRAS)G12C突变蛋白抑制剂MRTX849[1]、布鲁顿酪氨酸激酶(Bruton's tyrosine kinase,BTK)抑制剂[2]、血小板衍生生长因子受体β(platelet-derived growth factor receptor β,PDGFRβ)抑制剂[3]以及前列腺素合成酶(prostaglandin synthase,PGDS)抑制剂的研发等[4]。基于靶标结合口袋的水分子对于化合物优化的重要作用以及在SBDD中水分子的作用,已受到越来越多的关注和研究,如结合口袋水网络对于配体结合能力的影响[5]、替换水分子对于卤键的贡献等方面[6]。由于并不是所有结构中水分子的替换都是有利的,有时候替换水分子可能造成能量损失,化合物的活性减弱或选择性降低。对此一系列基于不同原理的水分子计算软件和方法,也被开发用于预测水分子的作用,如WaterMap[7]、3D-RISM[5]、SZMAP[6]等。在这些软件以及对于软件的测评中,也报道了一些药物设计的实例作为方法的验证[8-9]。然而,目前对于水分子的作用,特别是结合药物设计实例分析结构改造中替换水分子对于化合物的影响,还缺乏系统地总结介绍。
本文结合近年来药物设计中的代表性研究实例,较系统地对药物设计中基于替换水分子进行化合物优化的研究进行总结和展望,并对水分子和配体亲和力的关系以及预测水分子的软件和方法进行简要介绍,以期为基于结构的药物设计和新型药物分子的研发提供参考。
1 替换水分子指导药物设计的基本理论和方法
1.1 基本理论
绝大部分生物化学过程都在溶液中发生,在药物分子与靶标的结合过程中,除两者之间相互作用之外,溶液中的水分子状态也发生了改变。当药物和靶标发生结合时,一方面药物分子从溶液进入靶标结合口袋,另一方面结合口袋中原有的水分子被替换到溶液中,两者周围的水分子状态都发生了改变。
水分子对于化合物结合活性的贡献,可以从热力学的角度,根据化合物与靶标结合自由能的变化做出解释。化合物与靶标的解离平衡常数Kd,可以根据定义表示为化合物与靶标的结合自由能ΔG:
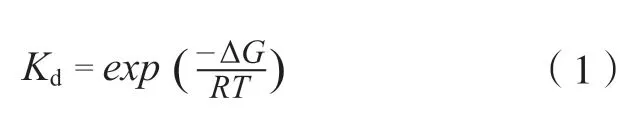
其中ΔG是结合自由能,R和T分别为气体常数和温度。ΔG越低,Kd越小,表明化合物与靶标的结合能力越强。由于ΔG是状态函数,根据热力学循环(见图1),ΔG分解为真空中化合物与蛋白的结合自由能变化ΔGvac,以及这一过程中溶剂的自由能变化ΔGsol:
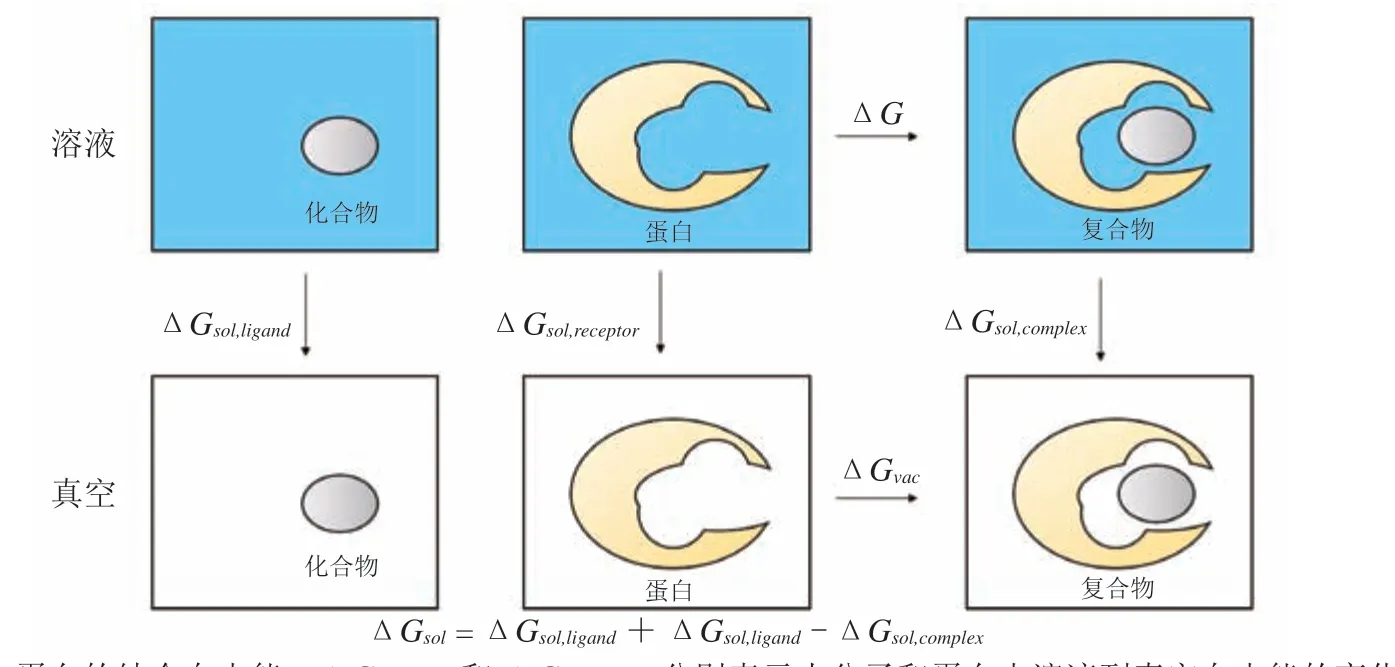
图1 化合物与靶标结合自由能的热力学循环分解示意图Figure 1 Schematic diagram of the thermodynamic cycle decomposition of the free energy of binding between a ligand and a protein

由此可以看出,ΔG不仅取决于化合物与靶标相互作用,也受到这一过程中溶剂水分子状态改变的影响。化合物的结构优化,可以简单看作是替换蛋白口袋中的水的过程。由于基团的引入,在增加与蛋白口袋相互作用的同时,也替换了蛋白口袋中的水。这一过程水分子总的贡献ΔGsol由各个水分子(i)的贡献加和而得到:

其中,ΔGsol,i是替换蛋白口袋中的水分子的自由能贡献。通过替换水分子i,ΔGsol,i将影响到化合物与蛋白的结合自由能,从而改变化合物与蛋白的结合活性。对于此部分水分子自由能的贡献,是药物设计过程中考虑水分子作用的核心。该项能量很难通过实验测量,通常只能通过理论计算得到。围绕该能量项有大量的相关理论计算研究,已经发展了一系列方法和软件,例如基于非均相流体理论(inhomogeneous fluid theory,IFT)的WaterMap等,此外还有基于其他计算方法的软件,如MOE的3D-RISM[5]、OpenEye的SZMAP[6]以及JAWS[10]等,将在本文进行简要介绍。值得注意的是,尽管许多软件可以重现晶体结构中的结构水,例如Aldeghi等[11]使用巨正则蒙特卡洛(grand canonical Monte Carlo,GCMC)方法对溴结构域蛋白(bromodomain,BRD)的大规模分析中发现,口袋中的结构水可以在计算中得到很好的重现,但由于晶体和计算条件环境的差异,以及计算方法的描述,两者是有所不同的,例如WaterMap实际上计算的是水位点,而非某个特定结构水分子的自由能。
1.2 设计方法
通过替换结合口袋中的水分子,或者与水分子形成相互作用,可以增强化合物结合能力、提高其选择性、改善其药动学性质。基于文献调研,笔者总结了常见的几种利用口袋中水分子的药物设计方法。如图2所示,当结合口袋小分子附近出现水分子时,有以下几种利用水分子改造化合物的方法:1)引入疏水基团,替换口袋中的水分子。口袋中存在不稳定的水分子,这些水分子通常位于口袋中的疏水区域,没有和周围的蛋白氨基酸残基形成相互作用,自由能较高,化合物替换这些水分子到溶液是自由能减小的过程(ΔGsol,i<0),有利于提高结合活性。这一方法已经在诸如KRAS G12C抑制剂MRTX849[1]、BTK激酶抑制剂[2]等多个化合物的结构优化中得到应用。2)引入如羟基等氢键给体或受体,替代水分子在这一位置的作用。当口袋中水分子与蛋白形成氢键,或者是与口袋中其他水分子构成稳定的氢键网络时,水分子能量较低,不容易被替换。替换这一水分子有可能造成能量损失,反而会减弱分子的结合活性(ΔGsol,i>0)。在PDGFRβ抑制剂[3]和PGDS抑制剂[4]的改造中,都发现了这样的例子。对于稳定的低能水分子,可以使用羟基等氢键给体或受体基团占据这种水分子的位置,同时与周围的残基或水网络形成氢键相互作用,替代这一水分子的作用,以增强或维持化合物的结合能力。另外,在酪氨酸蛋白激酶2(tyrosine kinase 2,TYK2)选择性抑制剂BMS986165的研究中[12],尽管替换水分子后化合物的活性没有得到显著提高,但改善了其药动学性质。此外,还存在一些情况,同源蛋白口袋周围残基的相似性很高,而口袋水分子的性质有所差异,这时通过水分子的替换可以提高化合物的选择性[13]。3)与水分子形成氢键相互作用。当口袋中水分子的自由能很低,替换水分子对结合不利时,也可以考虑不替换这一水分子,而是设计化合物与该水分子形成氢键。这种方式可以避免替换水分子造成的能量损失,也是一种利用口袋中水分子指导化合物改造的方法。例如在泛酪氨酸蛋白激酶(pan-tyrosine kinase,pan-TRK)抑制剂的研究中,通过与水分子形成盐桥而非替换水分子,起到了提高选择性的效果[14]。

图2 药物设计中利用口袋中水分子的策略示意图Figure 2 Schematic diagram of the strategy using water molecules in the pocket in drug design
在利用水分子指导化合物改造过程中,由于水分子自由能的高低无法单纯从观察晶体结构得知,理论计算成为了预测水分子位点及其性质的重要手段,目前已发展了多种方法和相关软件工具。这些方法和软件已经得到广泛应用,例如WaterMap已经在双特异性酪氨酸磷酸化调节激酶(dual-specificity tyrosine phosphorylation-regulated kinase,DYRK)[15]、β位淀粉样前体蛋白裂解酶1(β-site amyloid precursor protein cleaving enzyme 1,BACE-1)[13]、PDGFR-β[3]和TRK[16]等靶标指导其抑制剂的结构优化;JAWS已应用在p38α丝裂原蛋白激酶(p38α mitogen-activated protein kinase,p38α MAP)[17]、表皮生长因子受体(epidermal growth factor receptor,EGFR)[18]等的 抑 制剂 优 化中;SZMAP在周期蛋白依赖性蛋白激酶-1(cyclin-dependent kinase 1,CDK1)抑制剂的设计中解释了激酶选择性问题[11];3D-RISM在BACE-1高选择性抑制剂研究[11]和N9神经氨酸酶的研究中得到了应用[19]。
2 利用水位点进行药物设计
2.1 替换水分子增强结合活性
2.1.1 KRAS G12C抑制剂MRTX849的发现KRAS被认为是癌症中最常见的突变癌基因,在22%的癌症患者中均发现了KRAS突变,包括肺癌(17%)、结肠癌(33%)和胰腺癌(61%)。为了满足临床要求,需要直接靶向KRAS蛋白。然而,由于KRAS与内源性配体三磷酸鸟苷(guanosine triphosphate,GTP)具有较强的结合能力,蛋白表面较浅且结合域柔性较大,因此,KRAS曾一度被认为是“不可成药”的靶点[20]。直到近年来KRAS G12C抑制剂出现,为选择性靶向KRAS突变提供了一个新的方向。其中,Fell等[2]发现KRAS G12C共价抑制剂化 合 物1,其IC50为(4 400±73)nmol · L-1(见图3A),在结构改造过程中,晶体结构表明在结合口袋内,有1个水分子与Thr58侧链羟基以及Gly10羰基形成氢键(见图3B)。尝试在化合物1的哌嗪环上引入取代基,以替换这一水分子。当取代基为羟基甲基或羟基乙基时,活性并没有得到显著改善。当取代基为腈甲基替换水分子时,化合物1的活性提高了约100倍。进一步区分手性后发现,腈甲基S-构型异构体MRTX849(2)比R-构型活性高100倍,其IC50为(10±2) nmol · L-1。MRTX849与KRAS G12C复合物晶体结构表明(见图3C),腈甲基S-构型取代物替换了Thr58侧链羟基与Gly10羰基之间的水分子。这一水分子的替换,使得MRTX849的活性较化合物1提高了约400倍。

图3 KRAS G12C抑制剂MRTX849的发现过程Figure 3 Discovery of KRAS G12C inhibitor MRTX849
2.1.2 布鲁顿酪氨酸激酶抑制剂的设计BTK是非受体酪氨酸激酶家族的成员,在B淋巴细胞、巨噬细胞、中性粒细胞、树突细胞、红细胞、血小板和造血干细胞中表达。研究表明BTK是B细胞抗原受体(B-cell receptor,BCR)信号传导和激活的关键信号蛋白。抑制BTK有望治疗类风湿性关节炎等自身免疫性疾病[21]。武田制药公司研究人员通过基于片段的药物设计得到化合物3(IC50= 0.1 μmol · L-1)[3](见图4A),根据化合物3与BTK的复合物结构发现吡啶与P-loop上Phe413和Gly414氨基酸残基形成水分子介导的氢键作用(见图4B)。据此设计了一系列双环杂环取代该水分子。其中,吲哚取代的化合物4活性减弱(IC50= 850 nmol · L-1),而吡唑环取代的化合物5活性提高(IC50= 0.012 nmol · L-1)。在吡唑环的5位上进一步引入甲基,得到化合物6,IC50活性又提高至4 nmol · L-1。化合物6对淋巴细胞特异性蛋白酪氨酸激酶(lymphocyte-specific protein tyrosine kinase,Lck)的选择性达到了100倍以上,对RAJI细胞中BTK自磷酸化的抑制作用,化合物5和6细胞活性分别为92和28 nmol · L-1。从化合物6与BTK复合物晶体结构(见图4C)表明,吡唑环取代了水分子,环上的2个氮原子分别与Phe413和Gly414形成氢键相互作用。值得注意的是,相较于吡唑环取代的化合物5,吲哚取代的化合物4仅缺少1个氮原子形成的氢键,在替换水分子后活性不但没有提升,反而下降。当化合物为吡唑环时,替代水分子并形成氢键,才提高了化合物的活性。
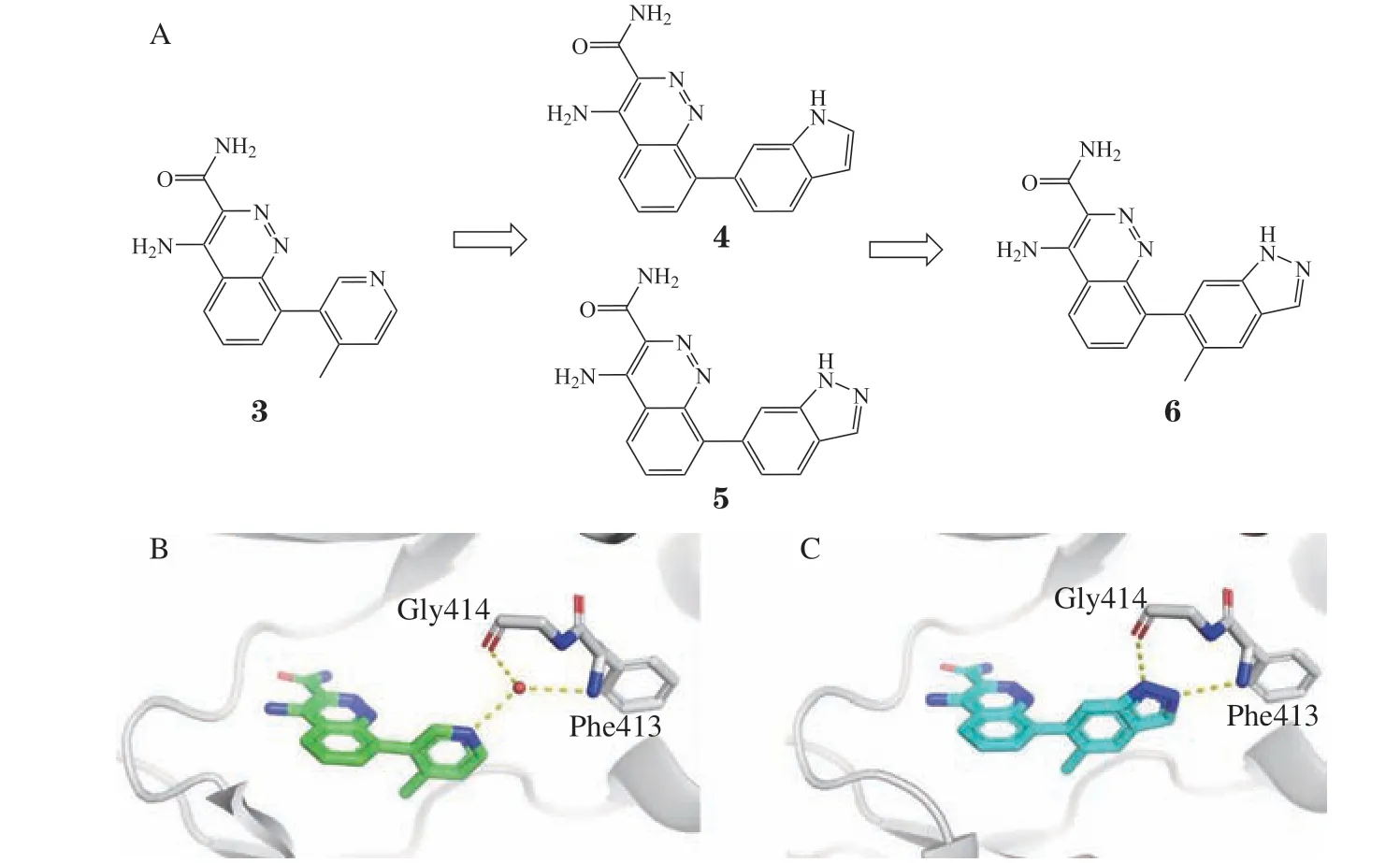
图4 布鲁顿酪氨酸激酶抑制剂的结构改造过程Figure 4 Structural modification of BTK inhibitors
2.1.3 血小板衍生生长因子受体β抑制剂的发现PDGFR是酪氨酸激酶家族中开发临床有效抑制剂的主要靶标之一。Horbert等[4]在PDGFRβ抑制剂化合物7的结构优化过程中,用同源模建方法构建了PDGFR激酶结构域的蛋白结构,通过分子对接研究先导化合物结合模式,并合成了化合物进行验证。在构效关系分析中,发现仅从分子对接结合模式,并不能解释化合物在PDGFR疏水口袋I(PDGFR hydrophobic pocket I,PDGFR HPI)的构效关系。研究发现,分析这种现象可以用取代水分子的热力学贡献来解释。通过WaterMap软件计算发现,在HPI有一系列的水位点尚未被利用(见图5A)。其中,位于苯环间位的3个水位点自由能,分别为5.5、9.9和25.8 kJ · mol-1,属于高能的水位点,而在对位的水位点是低自由能的水位点,其自由能为-15.9 kJ · mol-1。在化合物7的苯环上的间位和对位进行取代,尝试替换水位点。研究发现在对位引入氯或甲氧基得到化合物8和9,替换低自由能的稳定水位点,化合物8和9的活性反而下降,分别为31和228 μmol · L-1。当取代基为羟基得到化合物10,由于可以替代稳定水分子的作用,化合物10的活性提高了80倍,IC50为0.1 μmol · L-1。引入氯或甲氧基替换间位的高能水时,都可以起到增强活性的效果,化合物11和12的活性提高5 ~ 10倍。在对位引入羟基替代低能量的水分子,在间位引入甲氧基替换高能不稳定的水分子,优化得到化合物13,活性提高了40倍,IC50为20 nmol · L-1。综上所述,由于位于化合物邻位的水分子的自由能高,化合物的卤素或甲氧基取代后增加了活性,位于对位的水分子自由能低,只有被羟基替代,才能降低结合自由能,提升化合物的结合能力。
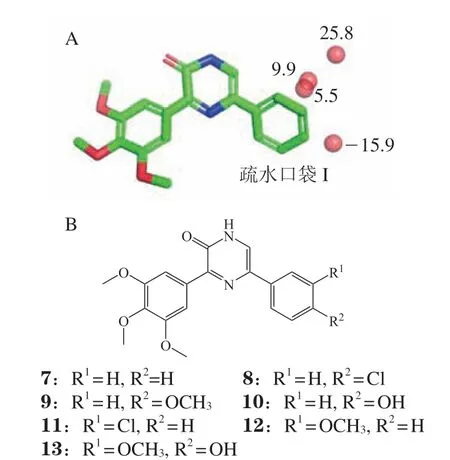
图5 血小板衍生生长因子受体β抑制剂的结构优化过程Figure 5 Structural optimization of PDGFRβ inhibitors
2.2 替换水分子增强选择性
阿尔茨海默病常见的病理特征是淀粉样β肽(amyloid β,Aβ)组成的淀粉样斑块,而Aβ是由BACE1和淀粉样前体蛋白(amyloid precursor protein,APP)产生[22]。BACE1是治疗阿尔茨海默病的极具前景的靶标之一。BACE2是BACE1的同源蛋白,研究表明BACE2在外周组织和大脑中有多种作用,长期抑制BACE2可能会引起一系列的副作用。然而,序列分析表明,BACE2与BACE1的序列一致性和相似性分别为55%和71%,配体结合口袋周围残基的一致性和相似性更是高达78%和88%,这给选择性抑制BACE1的 研发带来了难题。Fujimoto等[14]通过MOE软件溶剂分析模块计算,发现在BACE1中,Ser35和Tyr71间的区域有2个水分子能量较高,自由能分别为5.6和1.4 kcal · mol-1。然而在BACE2中,这2个水分子的自由能较低,分别为-2.3和-0.2 kcal · mol-1。依据此项计算,将二氢-1,3-嗪类化合物14进行头部改造(见图6A),在4位增加线性的丙炔基取代,得到化合物15,尽管其对于BACE1的活性略微降低,但选择性大幅提升,对BACE2的活性从166 nmol · L-1降低至1 778 nmol · L-1。化合物14与15与BACE1的复合物晶体结构表明,化合物15的丙炔基成功取代了S2’口袋中的1个水分子(见图6B和6C),除此之外并没有和氨基酸残基形成相互作用。尽管BACE1和BACE2在S2’口袋的残基类似,但其中的水位点自由能有明显差异,BACE2的水分子与残基紧密结合,属于低能水,而BACE1的结合比较松散,属于高能水分子。从水位点的自由能可以解释化合物15选择性大幅提高的原因。在后续的结构优化中,保留了丙炔基取代,将腈替换为氟甲氧基,并引入螺环以稳定生物活性构象,得到的化合物16,其对BACE1的IC50为24 nmol · L-1,对BACE2 的IC50为2 570 nmol · L-1,选择性从原来的5.2倍提高到107倍[14]。
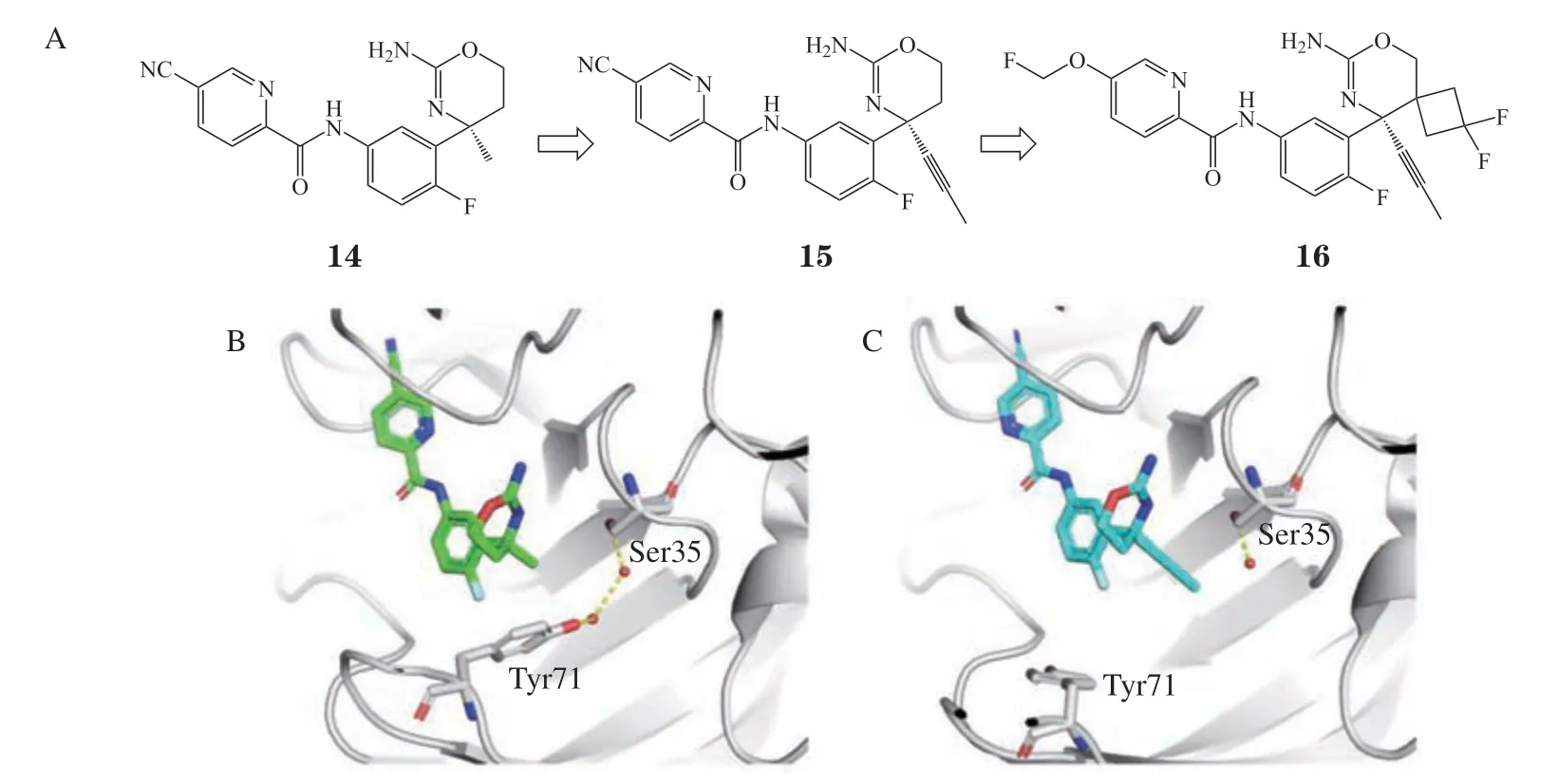
图6 β位淀粉样前体蛋白裂解酶1抑制剂的发现过程Figure 6 Discovery of BACE1 inhibitors
2.3 替换水分子改善亲脂效率
化合物17(见图7A)是酪氨酸蛋白激酶A(tyrosine kinase A, TRKA)变构抑制剂,对于TRKA具有良好的活性及选择性,但亲脂效率(lipophilic efficiency,LipE)仅为2.2,有较大的提升空间。Bagal等[17]通过使用WaterMap软件分析其共晶结构(PDB ID:6D1Y),发现在激酶的铰链区有一系列自由能较高的水分子,而在吡唑甲基位置,Arg738附近有一系列自由能低的稳定水分子(见图7B)。考虑以上2个因素,设计了7 000个分子进行分子对接,选择并合成了打分较高的100个分子并进行细胞实验,发现了化合物18的活性较17显著提高,对TRKA的IC50达到50 nmol · L-1,并保持了比TRKB/C高80倍的TRKA选择性。同时由于亲水基团的取代,油水分配系数增加(logD = 2.3),化合物18的亲脂效率得到了提高(LipE = 5.0)。化合物18与TRKA的共晶结构表明(见图7C),吡啶上的甲基被甲酰胺取代后,与侧链形成氢键相互作用,替换了Arg738附近稳定的水分子。
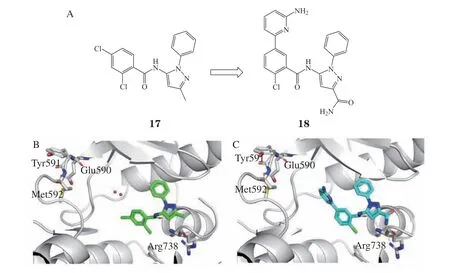
图7 酪氨酸蛋白激酶A变构抑制剂的结构改造过程Figure 7 Structural modification of TRKA allosteric modulator
2.4 通过替换水分子优化化合物药动学性质
酪氨酸激酶2(tyrosine kinase 2,TYK2)是JAK激酶家族(Janus kinase,JAK)成员之一,该家族还包括JAK1、JAK2和JAK3。JAK非受体酪氨酸激酶家族在介导引起炎症的多种细胞因子的信号传导中起关键作用。JAK激酶的小分子抑制剂有望治疗各种严重炎症和自身免疫性疾病。大部分JAK抑制剂都是与激酶的腺嘌呤核苷三磷酸(adenosine triphosphate,ATP)结 合 位 点JH1结构域结合,通过阻断ATP下游磷酸化,以及由此产生的信号通路转导来阻止激酶的催化活性[23]。化合物19(见图8A)选择性地与TYK2的假激酶(pseudokinase)JH2结构域结合,抑制TYK2的功能。这种别构位点的结合,使得化合物19在实现最大功效的同时,具有对于其他激酶的高选择性。化合物19对TYK2 JH2结构域有良好的亲和力(IC50= 0.5 nmol · L-1),对TYK2、JAK1、JAK2和JAK3的JH1结构域具有良好的选择性(IC50>10 000 nmol · L-1),对其他激酶家族成员也有很好的选择性。化合物19在进入临床前的结构优化过程中,研究人员通过分析化合物19与TYK2的晶体复合物结构发现(见图8B),化合物19的甲基砜占据了由Pro694与Leu741的侧链,以及Asn739主链形成的疏水口袋。在甲基砜的侧面,Gln597和Arg738之间有1个水分子,与化合物的N-氘代甲基甲酰胺羰基氧和砜基上的氧形成氢键。针对这一结构特点,尝试替换分子。设计思路是将氢键受体连接到砜基的邻位,替换水分子并与Arg738形成氢键相互作用。首先用甲氧基替换砜基得到化合物20(IC50= 5.2 nmol · L-1),尽管20活性比19低,但由于化合物20的极性表面积更小,是探索替换水更佳的起点。在化合物20的基础上,在甲氧基的邻位尝试不同取代基。当甲氧基的邻位是氰基、羧基、酰胺以及甲酰胺时,活性都得到了提升,与化合物19的抑制活性相当,但人全血(human whole blood,hWB)结合能力依然不高(IC50= 3 270 nmol · L-1)。尽管酰胺取代的化合物在后续的实验中发现微粒体稳定性差以及渗透性差,酰胺取代类化合物与TYK2 JH2晶体复合物的结构表明(见图8C),酰胺取代基通过与Arg738形成氢键,成功替换了Gln597和Arg738之间的结构水分子,且没有降低化合物的亲和力。在后续的优化中,继续保留了与Arg738的相互作用并优化与酰胺相关的微粒体稳定性及渗透性问题,最终得到化合物BMS-986165(21),在保持TYK2 JH2活性的基础上,与起始化合物19相比,细胞活性IC50提高了5倍;对hWB的IC50为13 nmol · L-1,活性提高了10倍[19]。

图8 酪氨酸蛋白激酶2 JH2抑制剂BMS-986165的发现过程Figure 8 Discovery of TYK2 JH2 inhibitor BMS-986165
2.5 替换水分子导致化合物活性或选择性下降
并不是所有水分子的替换都是有利的,有时候替换水分子也会带来相反效果,可能造成化合物活性的下降,或选择性降低。以下是2个替换水分子造成活性减弱,以及选择性降低的案例:1)研究人员对PGDS抑制剂进行结构改造,以替换口袋中的水分子,虽然晶体结构表明成功替换了结合口袋中的水分子,但活性测试却发现水分子的替换造成了化合物活性的显著降低。2)pan-TRK抑制剂对激酶家族的选择性不高,研究人员选择的是不替换水分子的策略,以提高抑制剂的选择性,如果替换口袋中的水分子,虽然化合物活性略微提高,但其选择性将大幅下降。
2.5.1 替换水分子减弱前列腺素合成酶抑制剂活性PGDS将环氧合酶(cyclooxygenase,COX)产生的前列腺素H-2(prostaglandin H-2,PGH-2)转化为前列腺素D-2(PGD-2),介导细胞的过敏和炎症反应。PGDS分为造血性前列腺素合成酶(hematopoietic PGDS,H-PGDS)和脂蛋白性前列腺素合成酶(lipocalin PGDS,L-PGDS)2种类型,选择性H-PGDS抑制剂被认为是潜在的抗过敏和抗炎药[24-25]。H-PGDS的晶体结构表明,抑制剂的结合口袋氨基酸残基Thr159侧链羟基与Leu199主链之间,有1个保守的水分子(w1),与抑制剂的氮形成氢键,w1与第2个水分子(w2)形成氢键,介导了第3个水分子(w3)与Ile155主链形成的盐桥并构成氢键网络(见图9)。辉瑞公司在化合物22的构效关系探索过程中(见图9A)[5],尝试在异喹啉环引入取代基,替换口袋中的水分子。随后在异喹啉的3位上引入甲基后,化合物23的结合活性较22略微降低,从原来的2.34 nmol · L-1变为8.26 nmol · L-1。从在化合物23与H-PGDS的晶体结构中(见图9B),甲基替换了水网络的水分子w2。将异喹啉环改为萘环并在原来氮的位置上加上取代基,以替换水网络的水分子w1。晶体结构表明,羟基甲基(化合物24)和氨基甲基(化合物25)均能够起到替换w1水分子的作用,替代原来的水分子与Thr159、Leu199以及w2形成氢键(见图9C和9D)。然而,尽管成功替换了水分子w1,化合物的活性却大大降低,化合物24的活性(IC50= 1 480 nmol · L-1)相较化合物22降低,化合物25的活性(IC50= 845 nmol · L-1)相较化合物22降低[5](见图9C和9D)。

图9抑制剂22~25与造血性前列腺素合成酶的构效关系探索Figure 9 Exploration of the structure-activity relationship of H-PGDS inhibitors 22~25
2.5.2 替换水分子导致泛酪氨酸激酶受体原肌球蛋白激酶抑制剂选择性降低由于慢性疼痛疾病的神经营养因子(nerve growth factor,NGF)通过TRK家族发出信号,TRK激酶抑制剂有望成为调节慢性疼痛的靶标[26]。辉瑞公司研发的化合物26[20](见图10A),对TRK家族具有良好的抑制活性,对TRKA、TRKB、TRKC的细胞活性IC50分别为6、2和1 nmol · L-1。但化合物26的选择性不佳,对于其他激酶也有结合,其激酶选择性基尼分数(Gini score,GS)为0.59。为了提高化合物的激酶选择性,设计减少化合物与铰链区的氢键作用。从晶体结构发现化合物26的氨基吡咯并嘧啶环与Glu590产生氢键相互作用(见图10B)。与TRKA未结合抑制剂化合物21的晶体结构对比(见图10C),发现化合物26氨基吡咯并嘧啶环上的氨基,替换TRKA蛋白中该位置的1个保守水分子(见图10C)。研究人员设计将氨基去除,不替换保守水分子而只形成1个由该水分子介导的氢键。去除氨基的化合物27,其抑制活性略微降低,而激酶选择性显著提高,GS提高到0.82。如果选择替换水分子,虽然可以略微提高化合物结合能力,但化合物激酶选择性将降低。

图10 泛酪氨酸激酶受体原肌球蛋白抑制剂的结构改造过程Figure 10 Structural modification of pan-TRK inhibitors
3 计算水位点性质相关方法及软件
发现口袋中水位点,并评价位点中的水分子是否适宜于被替换,对于利用替换水的方法进行SBDD至关重要。尽管蛋白口袋中的水分子位置可以从晶体结构中得知,但晶体结构中的水分子有时并不准确,特别是在分辨率不高的情况下,有可能只是假象或错误指认的电子密度[27-28]。随着计算方法的发展以及计算机性能的提高,许多计算方法都可以较为准确地放置水分子,一些软件在测试中已经可以与高分辨率的晶体水分子相符[10-12]。值得注意的是由于晶体与实际溶液状态的差异,一些计算软件预测的并不是晶体状态下水分子的分布。此外,仅从晶体结构,也无法判断出替换活性口袋中的水分子是否能够增强小分子的结合活性。
理论计算是预测水分子位点及其性质的重要手段,已发展了多种方法和相关软件工具。包括简单地基于晶体结构中的水分子进行评分、基于格点的取样方法以及基于分子动力学模拟的方法等。这些方法和软件的计算速度不同,计算准确度也各有差异,有着不同的适用范围。
其中,基于知识的方法直接通过评价晶体里水与周围环境的相互作用,预测该水分子是否容易被替换,其代表性的程序有Rossato等[29]开发的AQUARIUS、Raymer等[30]开 发 的Consolv、Verdonk等[31]开发的SuperStar以及Kellogg等[32]开发的WaterRank等。这些方法直接计算晶体结构中现有水的性质,但不能预测水位点的位置。
基于格点方法的程序通常使用格点来分析单个水放置的最佳位点,计算速度也比较快,代表程序有3D-RISM[15]、 SZMAP[33]、 WaterFLAP[34]等。其中3D-RISM根据液体在巨正则系综的密度泛函理论,在蛋白周围产生近似的溶剂分布,通过对自洽方程的评估给出水分子可能的位置。SZMAP在格点上计算泊松-玻尔兹曼的有效势,然后用单个水分子作为探针在不同方向上取样,通过这种方法来计算转动和平动熵的贡献。WaterFLAP使用格点分子相互作用场来定位水分子的位点,并使用一系列经验参数来计算水位点的能量。由于基于格点的方法把水分子近似为独立的粒子,并不尝试生成多个水的所有可能位置。基于格点的方法通常不需要耗费较大的计算资源,因此在平台软件以及当计算任务很多时被广泛采用。
基于模拟采样的方法是通过数值计算生成多个水分子所有可能的构象,并通过统计分析方法找到最低的能量以及位置,代表性的软件有薛定谔的WaterMap[35],通过将蛋白置于水盒子中进行分子动力学(molecular dynamics,MD)模拟,分析轨迹找出水位点,然后通过非均相溶剂化理论(inhomogeneous solvation theory,IFT)[36]计算这些水位点的自由能。除了MD模拟方法,还可以采用蒙特卡洛方法,如GCMC方法[37]、双重解耦-蒙特卡洛[38]等。基于模拟的方法优势在于可以描述出复杂的水分子网络,并解析出水分子的热力学性质。这个过程中需要MD模拟或者蒙特卡洛方法对水分子进行采样,因此需要耗费较大的计算资源,但有研究表明比前2种方法具有更高的准确性[16]。
4 结语与展望
通过替换靶标结合口袋中的水分子指导药物设计,近年来在一系列分子的结构优化中,已经成功地指导结构改造,提高了配体结合能力、选择性以及药动学等性质。此外,随着结构生物学、计算机科学的发展,通过蛋白质结构,预测水位点自由能的计算方法变得越来越准确快速,也大力推动了水分子替换这一方法在SBDD中的应用。
尽管利用水分子指导药物设计目前取得了一系列进展,但还有很大的发展空间。首先,目前大部分研究中对于口袋中的水分子考虑较少,在SBDD过程中仅考虑化合物和蛋白的相互作用。其次,仅晶体结构中发现水分子,而较少计算这些水分子的自由能性质,因此只能通过实验尝试不同类型的基团,尝试对水分子进行替换或形成相互作用。最后,利用计算软件预测水分子自由能,多是在结果分析用于解释化合物构效,前期用于指导化合物改造较少。总之,结合药物化学、结构生物学和计算机辅助药物设计等多种技术,利用口袋中的水分子进行药物设计,在未来新药研发中将发挥更为重要的作用。
