多位点关联分析方法学的研究进展
2022-01-27温阳俊冯建英张瑾
温阳俊,冯建英,张瑾,*
(1.南京农业大学理学院,江苏 南京 210095;2.南京农业大学作物遗传与种质创新国家重点实验室,江苏 南京 210095)
近年来,多位点模型(multi-locus model)在全基因组关联分析(genome-wide association study,GWAS)中引起了广泛关注[1-2],它在多重检验、群体结构和多基因背景控制方面都体现了一定优势。其中,以混合线性模型为框架的多位点GWAS方法,比如,多位点随机SNP效应混合线性模型(multi-locus random-SNP-effect mixed linear model,mrMLM)[3]、快速多位点随机SNP效应有效混合模型关联(FAST multi-locus random-SNP-effect efficient mixed model association,FASTmrEMMA)[4-5]以及全基因组复合区间作图(genome-wide composite interval mapping,GCIM)[5-7]能够进一步提高数量性状核苷酸(quantitative trait nucleotide,QTN)的检测功效,降低假阳性率,特别适合小效应和连锁QTN的检测。多位点方法在高粱[8]、水稻[9]、玉米[10-11]、棉花[12]、大麦[13]、大豆[14]和桃[15]等植物中已广泛应用,并挖掘出新的显著QTN及候选基因。为此,笔者对多位点关联分析方法学的研究进展进行综述。
1 多位点GWAS方法学的研究进展
1.1 GWAS混合线性模型方法学的建立
混合线性模型(mixed linear model,MLM)或线性混合模型(linear mixed model,LMM)是指同时含有固定效应和随机效应的线性模型,其中随机效应和误差具有方差,因此也称之为方差分量模型[16]。
2005年以前,GWAS以基于Case-Control数据和家系数据的算法为主。虽然有人建议采用MLM方法,但相关成果未见报道。为此,Zhang等[17]利用品种资源群体品种系谱、分子标记和数量性状表型信息,通过品种系谱计算品种间数量性状基因座(quantitative trait locus,QTL)的后裔同样(identity-by-descent,IBD)矩阵以及多基因背景的加性亲缘关系矩阵,建立了GWAS的混合线性模型方法。此方法把QTL效应看成随机效应,模型中包含了QTL效应方差、多基因背景方差和误差方差共3个方差分量。
在自然群体研究中,由于品种系谱信息缺失而导致结果不可靠。针对这一问题,Yu等[18]改进了Zhang等[17]的方法,提出由分子标记信息计算的品种间亲缘关系K矩阵代替品种间IBD矩阵,并引入群体结构Q矩阵。这称为关联分析的Q+K混合线性模型方法。它把QTN效应看成固定效应,模型中仅包含多基因背景方差和误差方差2个方差分量。
Kang等[19]建立了有效混合模型关联(efficient mixed-model association,EMMA)方法,被视作混合线性模型GWAS算法的黄金标准。对方差分量估计来说属于精确算法,即检测每一个SNP都需要重新估计多基因背景方差和误差方差的比值。它利用谱分解将似然函数和参数估计量都表示为特征值、特征向量、标记信息和表型值的标量运算形式,极大优化了似然函数的求解,速度比早期Yu等[18]提出的方法要快很多。尽管与现在的算法相比,速度不算快,但需要放在历史背景下进行比较。
1.2 GWAS混合线性模型方法学的发展
在MLM方法中,如果SNP标记数太多和群体容量太大,则计算的运行时间长。为了降低运行时间,提高QTN检测功效,涌现了许多以单位点(single-locus)分析为主的快速MLM方法。标准的单位点混合线性模型为:
y=Wα+Xβ+Zu+ε
(1)
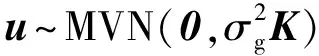
Zhang等[22]在Q+K的MLM关联分析基础上,将品种聚类成几组(压缩方法),用组间亲缘关系矩阵代替个体间亲缘关系矩阵,这称为压缩MLM(compressed mixed linear model,CMLM)方法;同时,在全基因组标记扫描时固定多基因背景方差与误差方差的比值,即P3D(population parameters previously determined)算法,它避免了多基因背景方差分量的重复估计,节约了计算时间。P3D对方差分量估计来说属于近似算法。后来Li等[23]从8种聚类算法(非加权组平均、非加权组质心、最长距离、可变类平均、加权的类间平均、加权的类间重心、最短距离和离差平方和)与3种类间亲缘关系算法(平均数、最大值和中位数)的24种组合中寻找最优组合,以实施CMLM方法。这称为增强CMLM(enriched CMLM,ECMLM),它能提高检测功效10%左右。
Kang等[24]同期也提出用P3D算法提高EMMA的计算速度,这称为EMMA eXpedited(EMMAX)算法。随后,Zhou等[25]提出了全基因组有效混合模型关联(genome-wide efficient mixed-model association,GEMMA)精确算法。GEMMA算法有3个特点:1)对亲缘关系矩阵只需1次谱分解。2)似然函数、待估参数的估计量及其涉及的一阶、二阶导数中的行列式、矩阵迹、向量-矩阵-向量乘积,都表示为标量运算形式。它显著提高了EMMA的运算速度,对群体容量较大的情形尤其突出。3)与EMMA相比,GEMMA不允许基因型数据有缺失,需要完整的或者imputed SNP。
Svishcheva等[26]整合了基于得分检验快速关联(fast association score test-based analysis,FASTA)[27]和基于混合模型与回归的全基因组快速关联(genome-wide rapid association using mixed model and regression,GRAMMAR)[28]2种方法的优点,发展了GRAMMAR-Gamma两步法。第1步,先估计衡量样本关系的群体参数以及GRAMMAR-Gamma因子,通过K矩阵变换得到新的表型值;第2步,提出一种含有GRAMMAR-Gamma因子的得分检验统计量,来检验每一个SNP标记是否与性状显著关联,其运算复杂度非常接近理论上的最小值。
Lippert等[29]提出了谱分解变换线性混合模型(factored spectrally transformed linear mixed model,FaST-LMM)。针对大规模数据集,它不是利用所有SNP构建遗传相似矩阵(genetic similarity matrix),而是均匀地随机抽取部分SNP构建真实亲缘关系矩阵(realized relationship matrix,RRM),并且这部分SNP数量小于样本个体数量,从而在保证没有信息损失的前提下,计算仍然准确,而运行速度却得到了显著提高。同时,它在检测所有标记过程中,只需1次谱分解;利用K矩阵的谱分解变换表型、标记信息向量以及协变量,可使变换后的数据不相关。
Listgarten等[30]发展了FaST-LMM-Select方法,通过线性回归获得每个SNP的P值,当遗传控制因子(genomic control factor)达到最小值时,确定P值作为阈值,选择满足此阈值的一小部分SNP及其附近(比如2 cM以内)的标记,不参与亲缘关系矩阵构建,再利用FaST-LMM方法进行检测。这种方法能够提高检测功效,节约计算成本。
Wang等[31]提出了SUPER(settlement of MLM under progressively exclusive relationship)方法,包括如下基本步骤:1)先进行单标记的一般线性模型和混合线性模型分析。2)将基因组划分为若干bin区间,每个bin区间选出最显著的SNP作为代表。3)用限制性最大似然方法确定最优的bin区间大小以及数目,选出潜在关联QTN来控制背景效应。4)检测每个标记时,去除潜在关联QTN中与待检测标记高连锁不平衡(linkage disequilibrium,LD)的SNP,再用FaST-LMM方法检测显著关联标记。这样通过剔除与待检测标记高LD的潜在关联QTN,避免亲缘关系矩阵对待检测标记的过度矫正,提高检测功效。
Loh等[32]假设多基因效应服从高斯混合分布,利用快速变分近似计算表型残差,凭借表型预测贝叶斯模型与经典关联检验方法有机结合的追溯(retrospective)得分统计量检验残差与检测标记间的相关性,这称为BOLT-LMM方法。它具有快速计算的特点,表现在:计算类似相关个体混合模型关联得分检验(mixed-model association score test on related individuals,MASTOR)[33]的拟似然得分检验统计量;利用类似于GRAMMAR-Gamma方法进行校正;利用预处理共轭梯度来估计方差分量,代替矩阵特征值分解。它也具有高功效特点,表现在:利用快速变分近似计算后验概率;利用LD得分回归校正检验统计量。
Jiang等[34]提出了基于混合线性模型全基因组关联(MLM-based GWA,fastGWA),它是基于MLM框架的GWAS工具,利用SNP衍生的主成分来控制群体结构,利用系谱信息或稀疏遗传关系矩阵来控制亲缘关系,对biobank-scale数据进行GWAS。它利用稀疏矩阵的Cholesky分解,提出了基于格子搜索求解限制性最大似然函数(fastGWA-REML)算法,代替了矩阵行列式和逆运算,采用近似GRAMMAR-Gamma得分检验统计量。模拟研究表明:fastGWA具有可靠、稳健和高效的特点。
上述的所有快速方法中,都是1次仅对1个SNP进行检验,属于单位点分析,且待检测SNP效应都是固定效应。
1.3 多位点模型方法的发展
虽然单位点分析应用广泛,但是复杂性状是由多个微效基因共同控制,所以单位点分析并不符合数量性状的真实模型。因此,多位点模型方法得到了快速发展。标准的多位点遗传模型为:
(2)
式中:y、W、α和ε同模型(1);p表示关联标记个数;Xi和βi分别表示第i个n×1关联标记基因型向量和效应。
多位点分析一般以惩罚压缩方法或贝叶斯方法或这两者相结合的方式为主,例如弹性网[35]、经验贝叶斯[36]和经验贝叶斯LASSO(empirical Bayesian least absolute shrinkage and selection operator,EBLASSO)[37]。
Cho等[35]提出的借助弹性网变量选择惩罚压缩方法构建多位点模型,是一种多步策略。第1步,对每一个SNP进行线性回归,得到部分显著的标记;第2步,对这些标记利用弹性网惩罚回归,进一步得到显著相关标记;第3步,利用bootstrap抽样,对上述显著的SNP进行显著性检验。
Lü等[36]提出了主效QTN、上位性互作和环境互作检测的经验贝叶斯方法。它将上述所有效应放入同一个模型中,利用Xu[38]提出的经验贝叶斯方法估计所有效应,并对非零效应进行似然比检验,达到同时检测主效QTN、上位性互作和环境互作的目的。若标记数目大于样本容量50倍,经验贝叶斯方法不能有效估计模型中包含的效应。为克服这一问题,Wen等[39]将具有3层先验分布、采用内积和外积迭代方式的EBLASSO算法[37]推广到部分NCII交配设计上位性关联作图中,以剖析杂种优势的遗传基础,构建的遗传模型同时包含加性效应、显性效应、加性×加性效应、加性×显性效应、显性×加性效应和显性×显性效应。
如果标记数量是样本容量的几百甚至数千倍时,上述几种方法将会失效。因此,应该考虑如何在多位点模型中有效减少待估效应数量。
Zhou等[40]将LMM和稀疏回归模型相结合,在贝叶斯框架下构建了多位点混合模型。假定每个SNP效应是服从2个正态分布的混合分布的随机效应,以满足稀疏性,即大部分SNP效应为零;同时利用亲缘关系矩阵控制多基因背景。利用马尔科夫链蒙特卡洛(Markov chain Monte Carlo,MCMC)方法,从观测数据中获得参数后验分布的近似样本,用来估计超参数。这称为BSLMM(Bayesian sparse linear mixed model)方法。由于稀疏性,其运行速度较快,能处理至少1万个体(每个体50万SNP)的数据集。Moser等[41]考虑了贝叶斯混合分布模型(Bayesian mixture model,BayesR),将SNP效应看成是随机效应,服从 4个不同数量级方差正态分布的混合分布,需要估计的方差分量数不多。经过几轮Gibbs抽样后,大约有500个效应最终保留在模型当中。计算时间过长是多位点贝叶斯方法的主要缺点。
Tamba等[42]提出多位点模型两步ISIS EM-BLASSO(iterative modified-sure independence screening EM-Bayesian LASSO)算法。第1步,利用SIS-SCAD[43]算法筛选潜在关联标记。其做法是:检测每个标记与数量性状表型值相关的显著性,选择概率P≤0.01的SNP进行SCAD[44]压缩估计,以筛选潜在关联的p1个SNP。在剩余的标记中,再用SIS-SCAD算法筛选出另一部分潜在关联的p2个SNP;第2步,将第1步筛选的p1+p2个潜在关联SNP标记放入同一模型,用EM-BLASSO算法[38]和似然比检验,获得与数量性状显著关联的SNP及其效应与贡献大小。此算法的最大优点是计算速度快和检测功效高。
Wang等[45]提出了稀疏贝叶斯学习(sparse Bayesian learning,SBL)方法。该方法采用坐标下降算法,在所有其他参数当前值的基础上,一次更新1个参数来估计标记效应。相比LASSO算法,它利用L2类型惩罚函数,可以处理超过100 000的样本量数据。同时,Wang等[46]还构建了压缩岭回归(deshrinking ridge regression,DDR)检验统计量,进一步提高多位点模型QTN的检测功效。
1.4 多位点GWAS混合模型方法的发展
植物数量性状一般情况是受少数主基因和大量微效基因控制,因此考虑品种间亲缘关系矩阵的多位点混合模型方法更符合其生物学特点。
Wang 等[47]将压缩算法自适应LASSO(adaptive LASSO)与线性混合模型LMM相结合,利用LASSO得到初始显著的q个SNP,对每个k(0—q),考虑多基因遗传背景,构建LMM,利用最大似然估计方差分量,获得协方差矩阵并进行特征值分解,得到矩阵变换,并变换原始模型,使变换后的表型彼此不相关,同时计算贝叶斯信息准则(Bayesian information criteria,BIC)、拓展BIC(extended BIC,EBIC)和赤池信息准则(Akaike’s information criteria,AIC)。对k重复以上步骤,以BIC、EBIC或AIC最小为准则,找到最优子集模型。
Segura等[48]提出了一种多位点混合模型关联分析方法(multi-locus mixed-model,MLMM),它利用所有遗传标记构建了亲缘关系矩阵,将向前、向后的逐步回归思想应用到线性混合模型中,在回归的每一步之前先对多基因背景方差和误差方差进行估计,然后利用广义最小二乘来估计SNP效应,进行F检验并获得P值。随后,将最显著的SNP作为协变量放入模型中进行下一步逐步回归,所有协变量的P值与方差分量一起重新估计。重复这一过程,直到符合终止条件。它利用Gram-Schmidt正交化过程,以及对协变量矩阵进行QR分解来提高计算效率。
Liu等[49]提出了固定与随机交替概率统一模型(fixed and random model circulating probability unification,FarmCPU),同时结合MLMM[48]和FaST-LMM-Select[30]的优点,交替使用固定效应模型和随机效应模型,尽量避免群体结构、亲缘关系和候选QTN间的混杂,达到降低假阳性率的目的。在固定效应模型中,一次仅检验1个标记,可能关联SNP作为协变量来控制假阳性率;从显著关联SNP中选取可能关联SNP来计算亲缘关系矩阵,并用随机效应模型进行预测优化。在随机效应模型中,计算该亲缘关系矩阵所解释的表型变异是否达到极大似然值,以此来防止固定效应模型的过拟合问题。固定和随机效应模型一直交替使用直到没有新的可能关联SNP进入模型中。
上述3种方法以及1.2节的所有快速混合线性模型方法都将待检测SNP效应视为固定效应。Goddard等[50]认为将SNP视为随机效应,构建随机SNP效应模型,其效果优于固定SNP效应模型。例如,随机模型可以把SNP效应压缩为零[3,51]。然而,Goddard等[50]并没有给出估计随机SNP效应的快速算法。
Wang等[3]提出了基于两步法的mrMLM算法。首先,对每一标记进行全基因组单标记扫描,以一种较为宽松的显著标准选择潜在关联SNP;其次,将所选择的潜在关联标记放入多位点模型,通过经验贝叶斯[38]估计和似然比检验获得显著关联的QTN。它结合了P3D、FaST-LMM的模型变换,检测标记的随机效应,并用固定效应和误差方差联合估计这些技术。计算机模拟和真实数据分析表明,与单位点模型及固定SNP效应模型方法相比,mrMLM的检测功效和参数估计值精度更高,假阳性率和假阴性率得到了有效控制。Zhang等[52]利用GEMMA[25]算法思想、矩阵变换以及Miller矩阵等式[53]来化简矩阵行列式和逆运算,提高了mrMLM[3]方法的运行速度,这称为FASTmrMLM算法。
Wen等[4]提出了FASTmrEMMA算法。它是一种快速高效多位点两步GWAS方法。第1步,将SNP效应看成是随机效应,且采用3种加速技术来减少运行时间。这3种加速技术分别是:1)对原始MLM进行特殊的矩阵变换,将多基因背景和误差变异变换成标准正态离差;2)固定多基因背景方差与误差方差的比值,对全基因组每一个SNP进行单标记扫描;3)检测标记信息矩阵的非零特征值数降至1个。第2步,对全基因组扫描概率P≤0.005的SNP放入多位点模型,利用经验贝叶斯算法[38]进行参数估计,利用似然比检验选出与目标性状显著关联的QTN。Wen等[5]进一步优化了FASTmrEMMA算法,利用Woodbury矩阵恒等式和性质,替换了特征向量的计算,适用于关联分析大群体,计算速度比原算法提高了至少60%,其他性能与原算法一致。该算法具有QTN检测功效高、精度高、运算速度快、假阳性率和假阴性率低的特点。
目前,FASTmrEMMA算法的矩阵变换已有5个拓展。Zhang等[54]将其他染色体多基因背景与误差转换成标准正态离差,然后通过最小角回归在目标染色体上选择与性状潜在关联的标记,最后将所有潜在关联标记放入多位点模型以检测显著关联QTN,这就是pLARmEB方法。与此类似的还有pKWmEB[55]、TSLRF[56]、FastRR[57]以及F2群体GCIM[7]算法。其中,pKWmEB算法结合了非参数检验Kruskal-Wallis;TSLRF算法结合了随机森林;FastRR算法结合了压缩岭回归DRR[46]检验统计量。
1.5 软件平台的重要发展
目前有很多GWAS软件包可供利用(表1)。这里仅对几种主流的MLM方法软件平台作简单介绍。
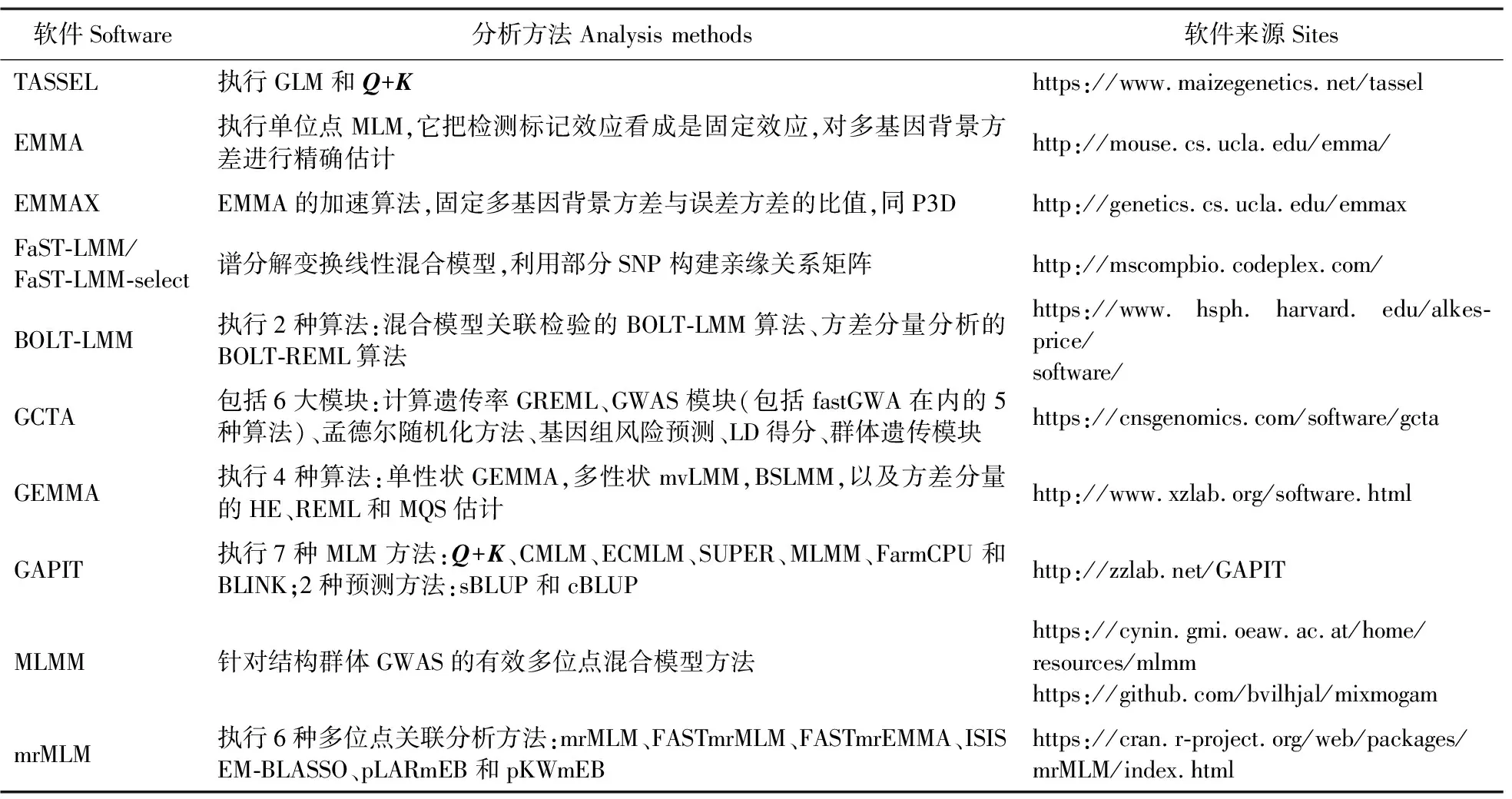
表1 全基因组关联分析混合线性模型方法常用软件平台Table 1 The common software platform for genome-wide association study(GWAS)based on mixed linear model(MLM)
TASSEL(trait analysis by association,evolution and linkage)[58]是一款早期的关联分析软件(https://www.maizegenetics.net/tassel),能够执行广义线性模型(general linear model,GLM)和MLM方法[18],有效控制群体结构和多基因遗传背景对关联分析的影响。
GEMMA(genome-wide efficient mixed model association)[25]是一款实现标准MLM的关联分析软件(http://www.xzlab.org/software.html),适用于大规模数据,计算效率高,可利用免费开源数字库。包括了 4种算法:1)单变量线性混合模型(GEMMA)[25],用于单性状分析,解释群体分层和样本结构,估计遗传率;2)多变量线性混合模型(mvLMM)[59],用于多性状分析,估计复杂性状之间遗传相关性;3)贝叶斯稀疏线性混合模型(BSLMM)[40],用于贝叶斯框架下的多位点分析;4)利用个体水平数据或汇总统计量(summary statistics)对方差分量进行HE、REML和MQS估计[60]。
GAPIT(genome association and prediction integrated tool)[61]自开发以来,目前已更新至v3.0版本(http://zzlab.net/GAPIT),可执行多种MLM关联分析方法,包括Q+K[18]、CMLM[22]、ECMLM[23]、SUPER[31]、MLMM[48]、FarmCPU[49]和BLINK[62]。同时还包括了2种预测方法:sBLUP和cBLUP[63]。
mrMLM v4.0[52]是章元明教授团队研发的R语言包(https://cran.r-project.org/web/packages/mrMLM/index.html)。它能够执行mrMLM[3]、FASTmrMLM[52]、FASTmrEMMA[4-5]、ISIS EM-BLASSO[42]、pLARmEB[54]和pKWmEB[55]6种多位点GWAS方法。
2 GWAS方法学研究的影响因素
2.1 假阳性率
GWAS面临着较高假阳性率(inflated false positive rates)的问题,这是因为群体结构(population structure)[64-65]、品种间的家系结构(family structure)和未知关联(cryptic relatedness)不准确[20],从而给GWAS带来了新的挑战。
目前,基于群体结构控制假阳性率的主要方法有基因组控制(genomic control,GC)[66]、结构关联(structured association,SA)[67]、主成分分析(principal components analysis,PCA)[68]和多维尺度(multidimensional scaling,MDS)[69]。这些方法能够较好控制群体分层,但是并没有考虑到所有品种的完整系谱,不适用于复杂的结构,例如品种间如果表现出复杂的亲缘关系,就不适合采用上述方法[48]。混合线性模型(MLM)方法能有效控制由大量微小效应多基因遗传背景和群体结构导致的偏差以及QTN检测的假阳性率,因此广泛应用于GWAS。
最近,Klasen等[70]认为群体结构校正过于严格,会影响真实关联的鉴定,从而提出了数量性状聚类关联检验(quantitative trait cluster association test,QTCAT)。该检验在考虑标记间相关性的同时,进行多标记关联。因此,QTCAT不需要进行群体结构校正,却比单标记方法更能反映出复杂性状的多基因特性。模拟数据分析表明QTCAT明显优于MLM方法[70]。
2.2 模型选择
2.2.1 单位点遗传模型的问题在GWAS中,单标记快速检测算法运算速度快,但它降低了检测功效。这是因为如果标记数量很多,利用单标记扫描算法,一次仅检验1个标记与性状间的关联,则需要进行Bonferroni校正,而这种校正过于严格,会导致一些小效应QTN达不到显著标准,即检测不到[3-4]。在基于混合模型的GWAS中,考虑了多基因背景控制,为了提高运行速度,每次对QTN进行单标记扫描,采用了固定多基因背景方差与误差方差的比值这种P3D算法,这是方差分量的近似算法。这无疑会使检测功效降低,不同群体降低程度有所不同。此外,当样本较大时,单标记扫描算法需要大规模求解矩阵逆运算,时间复杂度较高;并且对性状方差总的遗传贡献率估计困难。
2.2.2 多位点遗传模型的提出多位点模型比单位点模型更能解释植物复杂性状的遗传基础,它可以考虑相邻位点之间的潜在关系,具有更强的判别能力。在数学和统计上它是一种变量子集选择的过程,但它的难点是,可能的子集个数随SNP数的增加呈指数增长。若将海量标记全部放入同一多位点模型,则模型中变量(SNP数)远远大于样本容量,常用的线性回归分析无法解决这类问题[43]。虽然当前的压缩估计方法和Bayesian方法可以部分解决这类问题,但是以当前的CPU等硬件条件,运算量很大,运算时间很长。目前,惩罚压缩估计和Bayesian估计是多位点遗传模型的主流算法,有时也将这2种估计结合使用。但是,这2种方法很少有考虑到多基因背景或群体结构。大多数植物数量性状是由少数大效应和多数小效应基因所控制的,那么需要考虑多基因背景控制和群体结构的影响。对于微效QTN,采用多位点模型进行惩罚压缩估计,容易出现这些小效应都被压缩至零的现象[3]。不同模型的选择,会影响GWAS的结果。
2.3 计算与运行速度
数据量庞大带来了计算负担。除开发GPU等硬件设施之外,从算法本身进行创新与提速才是最重要的。充分利用降维方法,结合大规模矩阵分解与变换,是提高速度的有效途径之一。目前快速MLM方法的加速技术具体体现在:
1)单标记扫描事先固定多基因背景方差与误差方差的比率。CMLM[22]利用P3D技术加速MLM方法。EMMAX[24]利用P3D技术加速EMMA[19]方法。mrMLM[3]、FASTmrMLM[52]、FASTmrEMMA[4-5]都利用P3D技术加速。P3D技术对估计多基因背景方差和误差方差来说属于近似算法,它可以避免单标记扫描过程中重复估计这2个方差分量,从而提高计算速度。这是因为大部分SNP对性状不显著,因此在检测每一个标记时,这个比率近似相等。
2)化简大规模矩阵行列式和逆运算。MLM方法的似然函数或限制性似然函数对参数进行估计时都会涉及大规模的矩阵行列式和逆运算。比如,GEMMA[25]利用一次亲缘关系矩阵的谱分解,将行列式、矩阵迹以及向量-矩阵-向量乘积,都用标量乘积的形式表示出来。FASTmrMLM[52]采用GEMMA算法思想以及Miller矩阵等式[53],将逆运算化简成向量外积。FASTmrEMMA[4-5]利用矩阵特殊变换后,将矩阵行列式化简成向量内积运算以及一个向量-矩阵-向量乘法运算,二次型化简为特征向量的加权内积运算,单个标记信息矩阵的非零特征值只是一个正数(转换为向量内积运算)。利用Woodbury矩阵等式,将二次型进一步化简为与特征向量无关的向量内积运算。fastGWA[34]利用稀疏矩阵的Cholesky分解,提出基于格子搜索求解限制性最大似然函数(fastGWA-REML)算法,代替矩阵行列式和逆运算。充分利用数理统计知识、矩阵不同的分解形式等进行化简,化简成标量乘积、向量内积、向量-矩阵-向量乘法等运算,来提高运算速度。
3)采取有效的降维策略。在大数据时代,利用单一方法,比如单位点分析或多位点分析,有可能导致计算不可行或检测不准确。而采取多步降维方法是一种有效策略。比如,在进行多位点分析之前,可以将标记数量降到接近样本数量。由于LD使标记之间存在共线性,是高维数据的主要问题,可以利用LD进行SNP修剪[71],删除一些高度相关的标记,选择部分标记进行多位点分析。基于单倍型的SNP tagging[72]与SNP binning[51]也是高维数据降维的有效方式。此外,信念独立筛选(sure independence screening,SIS)[43]以及迭代信念独立筛选(iterative SIS)[43]算法可以处理超高维数据,它们主要利用标记与性状之间的边缘线性相关性来达到快速降维的目的。
3 小结与展望
近年来,多位点关联分析在剖析动植物的重要性状和人的复杂疾病中引起了学者们广泛关注,特别是以混合线性模型为框架构建的多位点GWAS方法,既能很好控制群体结构与多基因背景,降低假阳性率,又能提高小效应的检测功效。深入研究多位点GWAS方法不仅有助于深入了解复杂性状的遗传结构,还可以为分子设计育种和分子生物学研究提供新思路。
稀有等位基因(minor allele frequency,MAF<1%)检测一直是GWAS研究方法学的挑战[73]。在数据进行关联分析之前,通常是删除MAF<5%的位点,因为这些位点基本检测不到,此外由于Bonferroni校正,会导致阈值增加,其他显著位点统计功效降低[74]。但是没有理由认为稀有等位基因在生物学上不重要。事实上,由于纯化选择,许多有害等位基因都将以低频率出现[75]。目前,已有新的统计模型及软件用来分析人的稀有等位基因[76]。这为动植物数量性状稀有等位基因的挖掘提供了思路。
实际数据分析中,针对复杂性状的遗传结构和群体,如何选择最好的方法,并没有一个统一标准。不同GWAS方法往往会得到相似但不一致的结果[1-2,70]。例如某些显著QTN已经由一种方法检测到并通过功能验证具有生物学意义,但是其他方法却检测不到。这是因为利用了具有不同特征和优势的统计模型,比如单位点模型或多位点模型。我们建议用几种方法同时分析同一组数据,多种方法同时检测到的显著QTN应该是可靠的。同时,利用基因注释、表达、京都基因和基因组百科全书(Kyoto Encyclopedia of Genes and Genomes,KEGG)以及网络分析等现代组学手段,是比较容易发现显著QTN附近的可靠候选基因。
确定合适的显著性阈值一直是GWAS研究的热点[77]。在人的数据分析中,5×10-8是常用阈值。单位点分析中,Bonferroni校正是通用的方法,即0.05/p(p表示标记数)。但它假设了标记之间互相独立,并且对于作物遗传数据来说过于严格,以至丢失许多重要的小效应位点[1]。Xu[51]提出利用标记有效数来代替标记数p,修正了Bonferroni阈值。假发现率(false discovery rate,FDR)[78]也假设了独立性,使遗传区域的SNP由于LD具有相似的检验统计量,不太适合GWAS。置换检验被认为是建立显著性阈值的标准方法[79]。该方法在无效假设条件下,从检验统计量的分布中直接抽样。通过置换表型,同时保持基因型数据不变来计算每个标记的检验统计量。但该方法非常耗时,计算量庞大,不适合大数据的GWAS。同时,在MLM框架下,置换表型可能会破坏源于遗传关系的协方差结构[80]。多位点分析中,由于所有潜在关联标记的个数及效应能够在一个线性模型中同时确定并估计出来,所以无需进行Bonferroni校正[1-2]。可以设定LOD=3,对应的p阈值为0.000 2,比通用的0.05严格,能很好控制假阳性率,同时获得较高的统计功效[3-5]。这为确定合适的显著性阈值提供了方向。
随着其他类型的组学数据,如转录组学、蛋白质组学、代谢组学以及表观遗传学数据的开发利用[81-82],GWAS方法可以拓展到组学关联分析(omic-wide association study,OWAS)[83]。多组学变量在GWAS中被认为是性状的一种,可以在一定程度上弥补基因型和表型之间的未知关联,最终有利于选择性育种。例如,组学变量(在不同层次)被映射到与农艺性状相同的基因组位置,可以为复杂的遗传结构和潜在的生物途径提供多方面阐释。GWAS也能进行多性状分析,它可以利用个体水平的表型与基因型数据或者整合单性状GWAS分析的前期结果进行元分析[84]。在多组学关联分析研究中应用GWAS方法同时分析多水平和多性状,结合多组学的多位点关联分析方法能够为遗传学大数据分析提供新的途径。
