电催化氮还原合成氨电化学系统研究进展
2022-01-26刘恒源王海辉徐建鸿
刘恒源,王海辉,徐建鸿
(化学工程联合国家重点实验室,清华大学化学工程系,北京 100084)
引 言
氨是一种重要的化学品,是化肥工业的主要原料,其在国防、涂料、炸药等众多领域都具有广泛的应用[1-3],此外氨的氢含量可以达到17.6%(质量分数),且氨易于液化储运,是一种良好的储氢材料。2019 年,氨的全球产量为2.35 亿吨,是继硫酸之后产量第二高的商用化学品[4-5],在人类的生产生活中占据着重要地位。1909年Haber等提出工业氨合成法(即Haber-Bosch 法)实现了氨的大规模工业合成。该工艺的主反应方程式为:
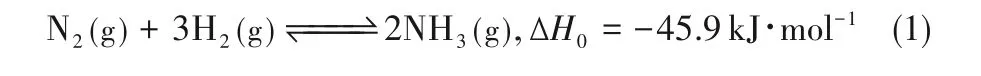
现行的Haber-Bosch 合成氨工艺(H-B 工艺)采用铁基催化剂,K2O、Al2O3作为催化助剂[6],工艺在高温(400~600℃)、高压(20~40 MPa)条件下进行[7]。H-B工艺的问世彻底改变了化肥的工业生产方式,从而革新了食物的供给模式,当今人体内约50%的氮原子来自这一工业过程[8]。由于H-B 工艺的固有效率高,总转化率可达97%[6],经过百余年的优化及改进,相关工艺流程已较为成熟,至今该工艺仍是工业合成氨领域中的主流生产工艺,全球范围内90%以上的氨是通过H-B工艺从化石燃料中产生的[1]。
然而,H-B 工艺面临着能耗问题和环境问题,维持高温、高压条件需要消耗巨大的能量;驱动反应的能量(>600 kJ•mol-1)大部分来自化石燃料,反应原料氢气一般由天然气蒸汽重整过程产出[9],归根溯源氢气的供应也来自化石燃料的使用,因而该工艺过程的进行将排放大量的温室气体,对环境造成严重影响。H-B 工艺的能耗高达全球能源总消耗的1%~2%[10-11],且每生产1 kg 的NH3即会产生2.16 kg 的温室气体,工艺产生的CO2占到全球总排放量的1.5%[12],随着时代的进步与发展,节能降耗、绿色环保成为生产生活中的重要趋势。H-B 工艺的能耗及环境问题日渐尖锐,因而现有诸多研究致力于绿色可持续性的合成氨工艺路线的开发。如图1所示,相关研究中,可行性较高的研究路径包括酶催化、多相催化、电催化、光催化等。其中电催化路径具有潜在的性能优势,前景广阔,是近年来氮还原合成氨领域中众多研究人员关注的热点所在。
1 电催化NRR工艺概述
电催化氮还原反应(电催化NRR)是替代传统H-B 工艺的可行性路径中最具发展潜力的工艺,具有节能低耗、绿色环保的特点,能够有效克服传统工艺的弊端。该过程的总反应方程式为:

相较于H-B工艺,电催化NRR合成氨工艺的优势体现在以下几方面:(1)H2O 替代H2作为氢源,减少了化石燃料的使用,原料无毒无害、来源广泛;(2)电催化NRR 可通过调节工作电位来打破反应的热力学限制;(3)从热力学上进行理论计算,电催化NRR 工艺比H-B 工艺节能20%[6];(4)反应条件温和,减少能耗,工艺过程的安全性高;(5)采用电力驱动,可以选择太阳能、风能、地热能等可再生的绿色能源作为电力供应;(6)工艺装置简单、操作灵活,无须昂贵的基础设施建设,可实现在偏远地区的合成氨工厂建设,实现氨的按需制备和精准制备,灵活性和可调谐性远远超过H-B 工艺[6];(7)电催化NRR 工艺相当于从潮湿的空气中合成氨,是一种低碳的合成方式,与时代需求“碳达峰、碳中和”极为吻合。综上所述,电催化NRR 工艺过程无论从原料、能源供给、绿色环保还是生产灵活性等方面都具有明显的优势,其有望成为替代传统H-B工艺的新型工艺路线。
衡量电催化NRR过程的性能一般采用产氨速率rNH3和法拉第效率FE(%)两个参数,前者表征活性,后者表征选择性。由于催化剂活性有限、阴极析氢反应竞争激烈等原因,现有大多数研究所能实现的产氨速率和法拉第效率仍然十分有限。根据美国能源部的ARPA-EREFUEL计划,电催化NRR工艺商用化所需要达到的性能目标为:在300 mA•cm-2的电流密度下,氨收率达到10-6mol•s-1•cm-2,法拉第效率需要达到90%,同时要求催化剂具有较高的稳定性,即在电解1000 h后其催化效率仅能够下降0.3%[13]。与此商用化标准相比较,目前大多数电催化NRR 研究所能够实现的性能远远不能够满足商用化标准,实现其工业化与商业化的路仍然漫长,需要大量深入的研究与突破。
2 电催化NRR过程分析及优化途径
2.1 电催化NRR电极反应过程及技术难点
氨的合成发生在阴极还原反应中,此电极上的界面反应如图2所示,主要分为三个步骤:N2的溶解和扩散,N2在电极表面的吸附、活化及加氢过程,NH3在电极表面的解吸。

图2 电催化合成氨阴极反应过程Fig.2 Schematic diagram of cathodic reaction process of electrocatalytic synthesis of ammonia
常温常压下N2在水中的溶解度较低,仅为0.66 mmol•L-1[14],作为反应原料,N2溶解及传质扩散过程的限制将显著影响整体的合成效率。N2的吸附及活化加氢过程是最为关键的步骤,N2的吸附活化过程主要有解离机理、缔合机理(交替配位、末端配位)、酶途径以及Mvk。前三种较为常见,详细过程如图3 所示。解离机理过程为N2首先解离为N原子吸附在表面,每个N 原子加氢形成NH3并从表面脱附以此完成催化过程,H-B 工艺过程即属于解离机理[15]。缔合机理(末端配位)中N2以分子形式吸附在表面,两氮原子间保持化学键连接,加氢首先发生在离表面较远的氮原子上,完成加氢后N―N 键断裂,外侧脱去一分子NH3,内侧N 原子开始进行氢化,形成一分子NH3后从表面脱附。缔合机理(交替配位)的前期过程与末端配位机理类似,但交替配位机理中两个N 原子交替进行加氢过程,率先完成加氢的N 原子形成NH3从表面脱附,剩下的氮原子继续进行加氢过程。两种缔合机理中,随着加氢过程的进行,两个氮原子间的化学键会有所减弱,N―N 键的断裂相对容易很多。因而相比于解离机理,缔合机理能耗更低,更容易进行。催化剂种类、结构以及反应条件不同时,氮还原合成氨所经历的反应过程也不尽相同,应用机理时需要具体情况具体分析。此外,还有酶途径、Mvk 两种机理,前者主要出现在固氮酶的氮还原反应中,该机理中N2仍然以分子的形式吸附,但两个N 原子没有远近关系,并列吸附在表面,加氢过程与交替配位机理相同[11]。Mvk 机理更多出现在过渡金属氮化物的催化过程中[16]。

图3 氮还原合成氨表面反应机理[15]Fig.3 Surface reaction mechanism of ammonia synthesis by nitrogen reduction[15]
在不同机理中,N2的活化均为决速步骤[17]。N2分子中的氮氮叁键具有较高的键能(941 kJ•mol-1),其活化及断裂的难度较大,使得N2分子在催化剂表面的吸附活化较为困难,造成电催化NRR 产氨速率不高,这是该领域中所面临的主要技术难题之一。另外,阴极存在析氢反应(HER)主导的竞争反应,影响反应的选择性,该过程中的电极反应及对应的电极电位见表1。阴极上的氮还原反应的电极电位与析氢反应接近,同时质子比N2更容易活化,从平衡、动力学两个角度衡量,析氢反应都更容易发生,因而提升工艺的法拉第效率难度较大。

表1 电催化NRR阴极电极反应及电极电位比较[18]Table 1 Electrode potential comparison of electrocatalytic NRR cathodic electrode reactions[18]
N2的加氢过程完成后生成的氨分子需要从表面脱附,此过程需要催化剂具有适中的N 原子吸附能力,吸附过弱则不能有效吸附和活化N2,吸附过强则导致产物不能从表面脱附。因而催化剂可以依据N 原子吸附能的火山形曲线进行选择,如图4所示。

图4 后过渡金属电催化氮还原的火山形曲线[19]Fig.4 Volcano plot for electrocatalytic nitrogen reduction on late transition metals[19]
目前电催化氮还原合成氨工艺所面临的主要技术难点有:(1)N2为惰性分子难以吸附、活化;(2)阴极存在析氢(HER)竞争反应,影响法拉第效率;(3)常温、常压下N2在水中的溶解度很低,传质影响较大;(4)反应过程对催化剂的N 吸附能力要求较高。从中可以总结出该领域中的几点关键科学问题:(1)N2分子的活化及合成氨的反应机理研究;(2)合成氨催化材料的构效关系研究;(3)氮气溶解及传质动力学研究与强化。
2.2 电催化NRR性能优化措施
现有研究即针对电催化NRR 领域中的关键技术难点,开发不同的优化及改性措施,如图5 所示,其主要可以分为两个方面:围绕催化材料的设计及改性;围绕电催化系统的修饰及改进。
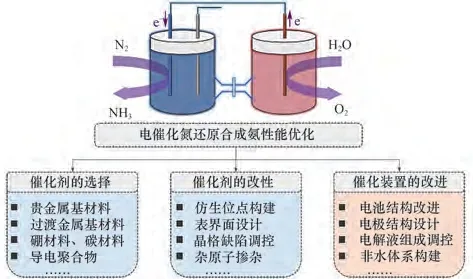
图5 电催化氮还原合成氨工艺性能优化策略Fig.5 Optimization strategies of electrocatalytic NRR process
催化材料的相关研究中,首先需要选择、设计并合成出合适的催化剂,而后需要对其进行改性以提升其催化性能。研究中采用的催化剂种类主要有:贵金属基材料(Ru[20]、Rh[21]、Au[22]等)、过渡金属基催化剂(Fe[23]、Co[24]、Ni[25]、Mo[26]等)、主族金属基材料(Bi[27]、Sn[28])、金属氧化物(Fe2O3等)、非金属基材料(硼材料[29]、碳材料[30]、导电聚合物[31])等。在催化剂形式的设计过程中,可采用仿生策略、态密度(DOS)分析以及密度泛函理论(DFT)计算等措施对催化剂的结构进行合理设计并对其催化性能进行理论预测。另外,常用的催化材料改性措施包括:形貌尺寸调控、晶格缺陷构建、表界面设计、杂原子掺杂、空位工程等以及几种措施间的协同效应[32-33]。催化剂的设计及改性是实现氮气高效活化的有效措施,因而也是本领域研究的重点所在。
然而,现有研究更多地围绕催化材料的相关问题进行展开,而对于电催化装置体系的改进研究相对较少,但对于电催化NRR 工艺的性能提升,二者同等重要,缺一不可。因而下文将重点综述电催化NRR的装置体系相关改进的研究进展。
3 电催化NRR 装置系统的改进及优化
3.1 电催化装置结构及优化
根据电池结构的不同,电催化反应装置可以分为四种类型[34]:背对背型、聚合物电解质膜型、H 型以及单槽型(图6)。
背对背型电解槽结构如图6(a)所示,中间为阴阳两极,两侧为阴极室和阳极室,两极间由膜隔开,膜可选用质子交换膜(PEM)或阴离子交换膜(AEM),研究采用前者居多,且一般采用Nafion 膜,该膜能在室温下高效传递质子。Lan 等[35]选用Nafion 211 膜,并用Li2SO4溶液进行充分离子交换,制备出H+/Li+混合传导膜,研究中发现,Li+的引入一定程度上能够抑制质子的传递过程。Renner等[36]尝试将AEM 引入本领域,并在低温、低压的条件下实现了电催化氨合成,但是其性能不及Nafion 膜的相关研究,因而研究中采用的仍主要为Nafion膜。

图6 温和条件下电催化NRR不同电池结构[34]Fig.6 Schematic diagram of different cell structures of electrocatalytic NRR under mild conditions[34]
聚合物电解质膜型(PEM型)电解槽结构如图6(b)所示,该结构中阳极室采用水溶液作为电解质,可引入参比电极更加精准测定体系的电极电位,同时隔膜与水溶液接触能够保持湿润状态,保证其较高的离子传导能力。Lan 等[37]使用Nafion 膜作为固态电解质,实验中在阴、阳两极室中均检测到氨,即存在阴极生成的氨透过Nafion 膜向阳极室的穿透现象,此现象的存在将导致还原产生的氨又转移至阳极被氧化,造成产氨速率降低、电能的浪费等问题。Chen 等[38]为了解决氨在膜两侧的穿透问题,将PEM型反应器结构进行改进,如图7所示,在催化剂层与Nafion 膜之间增加了气体扩散层,有效解决氨穿透问题的同时,限制了质子的传输途径,一定程度上抑制了阴极析氢反应的发生。

图7 改进后电催化氮还原合成氨三相反应器原理[38]Fig.7 Schematic view of the improved design in the threephase reactor for electrochemical ammonia synthesis[38]
H 型电解槽的结构如图6(c)所示,现有研究多采用此种结构,该结构中左右两侧为装有电解液的阴、阳两极室,中间由Nafion膜隔开。装置采用三电极体系,对电极处于阳极室中,工作电极与参比电极均置于阴极室中,由此可以更加准确地测定阴极电位。Chen 等[39]采用图8 所示的典型三电极装置,以Li+修饰的PEBCD 作为催化剂进行合成氨实验,实现了1.58 μg•h-1•cm-2的产氨速率及2.85%的法拉第效率。Liu 等[40]采用H 型电解槽,饱和甘汞电极作为参比电极,碳棒作为对电极,催化剂选择原子级超薄铑纳米片,实现了23.88 mg•h-1•(mg cat)-1的产氨速率。H 型电解槽中两极室间采用Nafion 膜隔开,使得阴阳两极所处环境相对独立,从而可以对两极室分别进行更灵活的条件控制。
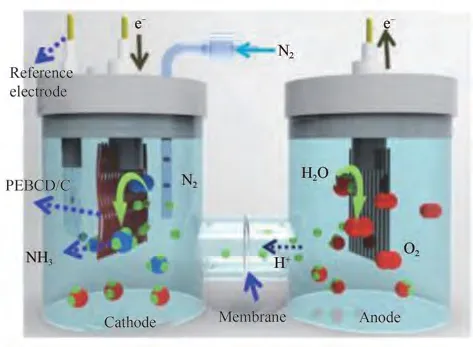
图8 电催化NRR H型三电极反应装置[39]Fig.8 Schematic diagram of electrocatalytic NRR H-type three-electrode reaction cell[39]
单槽型电解槽的结构如图6(d)所示,与H 型电解槽不同,单槽结构中阴、阳极室连通,两电极处于同一电解质溶液环境中。Köleli等[41]采用单槽结构,将聚吡咯电极作为工作电极,在0.1 mol•L-1Li2SO4/0.03 mol•L-1H+的溶液环境中实现了电催化NRR。Kugler 等[42]亦采用单槽结构的反应器,将Rh、Ru 沉积在随机结构的Ti 材料上并作为反应的催化剂,实现了常温下氨的电催化合成,说明单槽型反应器用于电催化NRR体系的有效性。
H 型和单槽型反应器是目前电催化NRR 研究中应用较多的两种电解槽形式,单槽结构虽然装置简单,易于操作,但由于阴、阳两极室处于连通状态,阴极上生成的氨分子容易扩散至阳极发生氧化,造成氨收率低、电能浪费等问题。而H 型电解槽采用膜将两极室隔开,离子在极室间选择性交换,两极相对独立,对于电极环境的调控更加简便、灵活,因而在这些方面,H型电催化反应装置优势更加明显。电解槽的结构及形式决定了电极和电解液的排布方式,一定程度上间接决定了体系中的物质传递路径以及反应场所的位置,因而在实际应用环境中需根据具体体系的特性对电解槽的形式及结构进行合理选择及设计。
3.2 电极结构的优化策略
现有研究一般采用三电极体系,即工作电极、参比电极和对电极,体系结构见图5和图8。参比电极一般选择电极电位相对稳定的Ag/AgCl、甘汞电极等,引入参比电极能够更加准确地测出工作电位;对电极一般选用铂电极、石墨电极等惰性电极,其仅为阳极氧化反应提供场所。工作电极一般由催化剂负载于集流体上制成,因而工作电极的设计及优化研究大部分是催化材料的合成及改性,但除此之外也可以对电极本身结构、组成及表面状态进行改进。
电极结构方面,有研究采用气体扩散电极以强化原料气的溶解及传质过程[43],其结构设计如图9所示。电极材料一般采用碳纤维骨架,如图9(a)所示,由于其疏水特性,产生了明显的气-液-固三相界面,为氮还原合成氨反应的发生提供了有利场所。然而,当电解质采用非水体系时,如图9(b)所示,电解质会深入渗透进碳纤维结构中,淹没电极,阻碍气体扩散,不利于气-液-固三相界面的形成。为解决此问题,研究将碳纤维骨架替换为不锈钢网,同时在气体一侧施加一定压力,如图9(c)、(d)所示,维持了三相界面,使电化学反应能够高效进行。

图9 电催化合成氨气体扩散电极结构的设计[43]Fig.9 Gas diffusion electrode design for ammonia electrosynthesis[43]
从类似的研究思路出发,为强化电极处气-液-固三相界面,Liu 等[44]对阴极结构进行设计,采用亲气型超薄多孔Bi5O7I 纳米管和亲水碳球构建了亲气-亲水异质结构电极,同时向电解质溶液中加入带正电荷的Bi5O7I 纳米管作为共催化剂,实现了高效电催化NRR 过程。此电极结构兼具亲水、亲气特性,能够有效提高催化剂表面的N2浓度,抑制析氢过程,促进反应产物的释放,有效强化了电催化过程,研究实现了31.46 mg•h-1•m-2的氨收率以及13.42%的法拉第效率,强化效果明显。
电极界面是电化学反应发生的主要场所,因此有必要对电极结构进行合理设计,为气体扩散提供有利空间,促进气-液-固三相界面的形成,保证反应的高效进行。
电极组成及表面状态方面,Chamoun等[45]研究发现,在铁电极中掺入铋或锡酸盐添加剂能够有效抑制水介质体系的HER,提高反应的选择性。Singh 等[46]定性分析了H2/NH3的选择性问题,研究表明当催化活性位点附近的电子或质子浓度较高时,析氢反应占主导地位,因而降低催化电极附近的电子或质子浓度理论上能够显著提高反应选择性。由于电极与外电路直接相连,因而对于电子的控制难度较大,对于质子的控制相对更加容易,因而可以在电极表面包裹一层非质子的疏水保护层,在允许N2通过的同时动态减缓质子向电极表面的扩散过程,从而适度降低电极附近的质子浓度,提升反应选择性;电子的控制方面,可以构建“金属绝缘体-催化剂”形式的电极,控制电子从电极穿过绝缘体到达催化剂金属层的隧穿,减缓电子生成和转移的速率,若电子的生成及转移过程变为决速步骤,则有望使得反应的选择性实现新的突破[12]。此类研究可以对反应的机理进行进一步挖掘,依据机理对电极的组成及表面状态进行合理改性,从而改善电子及质子的传递途径,实现对于反应活性及选择性的优化。
3.3 电解质溶液的优化策略
电解质溶液是物质传递、离子传导等过程发生的主要场所,现有研究中大多采用水溶液作为电解液,电解质溶液环境对于产氨速率、法拉第效率等性能会产生明显影响,因而有必要对电解质的组成、配比、pH等参数进行探究及优化。
无论是酸性还是碱性环境中,电催化NRR 的电极反应均涉及H+或OH-,因而电解液pH会对电极反应产生影响。Chen 等[38]采用Fe2O3-CNT 作为催化剂,探究了电解液在不同pH 条件下的电催化性能,pH=13.7、9.4、7.0、0.6 四组条件下的电流密度分布、平均产氨速率及法拉第效率如图10所示,酸性环境中电流密度最大,过渡到碱性环境时,电流密度下降,原因主要为随着pH 的增大质子浓度降低,且主体迁移对象由H+变为H2O。pH 对于产氨速率的影响并不明显,但法拉第效率随着pH 的增大有了明显提升,碱性环境中质子浓度低,析氢反应受阻,使得碱性环境中的法拉第效率普遍高于酸性环境。

图10 不同电解液中催化剂的电流密度分布、平均产氨速率及法拉第效率[38]Fig.10 Current density profile,average ammonia formation rate and Faradaic efficiency of catalysts in different electrolytes[38]
N2的传质、质子的传递过程对于电催化NRR 过程至关重要,二者与电解质环境有着密切的联系,因而可以通过调节电解液的组成及配比,一方面增强其对N2的溶解能力,打破传质限制,提高反应活性;另一方面可调控电解液中质子的传导方式,实现对HER的抑制,提高反应的选择性。Hao等[47]的研究表明,电解液中K+能够稳定关键中间体、抑制质子由本体溶液向电极表面的迁移,从而选择性削弱析氢反应,提升法拉第效率。Malkhandi 等[48]研究发现,利用碱性电解质中的有机硫化合物作为空间位阻有助于抑制HER的发生。Kim等[49]发现相较于纯水体系,2-丙醇对N2的溶解能力更强,且对HER具有一定的抑制作用,2-丙醇与水体积比9∶1时的混合体系对HER抑制效果更加明显,但是由于2-丙醇在还原环境中并不稳定,该研究所实现的产氨速率为1.54×10-11mol•s-1•cm-2,法拉第效率仅为0.9%。Kim等[50]采用H型电解槽,将0.1 mol•L-1LiCl/EDA(乙二胺)作为阴极电解液,0.05 mol•L-1H2SO4作为阳极电解液,在1.8 V的电池电压下电解1 h产生了7.73×10-7mol NH3,法拉第效率能够达到17.2%,并且研究证明EDA是电催化NRR 领域中性能优越的溶剂。Zhang 等[17]研究证明,N2在磷酸盐溶液中的溶解度比在其他水溶液中的溶解度高,将磷酸盐作为电解质可以强化N2的溶解及传质过程,从而提升产氨速率,采用K3PO4作为电解液,则K和PO3-4能够对NRR起到协同促进作用。
电解液作为物质传递、离子传导的主要场所,其组成、配比、酸碱度以及温度等参数对于电化学反应的进行至关重要,因而研究有必要对电解液的相关参数进行条件优化。
3.4 非水体系的构建
针对水溶液体系存在氮气溶解度低等问题,有研究试图开发非水体系,如离子液体等,意在为NRR 提供更有利的反应环境。Zhou 等[51]采用同时具备疏水特性及高氮溶解度的[P6,6,6,14][eFAP]离子液体作为电解液,并实现了60% 的法拉第效率。Suryanto 等[52]将非质子氟化溶剂与离子液体的混合物作为电解液,抑制质子迁移的同时提升N2的溶解度,从而实现产氨速率和法拉第效率的提升。
特别地,Li介导的电催化NRR 反应体系在产氨活性及选择性提升方面具有较大的发展潜力,近年来成为该领域中研究热点。该体系中的物质循环如图11 所示[53],Li+首先被还原为金属锂,而后金属锂与溶解在溶剂中的N2发生反应生成氮化锂(Li3N),此时Li3N 与质子载体提供的质子结合,反应生成NH3,反应方程式如式(7)所示,生成的NH3即溶解在溶剂中。在此过程中,质子源即为发生在阳极的H2或H2O 的氧化过程,质子产生后需通过质子载体将其从阳极运输至阴极并参与式(7)所示的反应,研究中一般采用乙醇作为质子载体,通过其在阴阳两极间进行EtOLi 与EtOH 反复的结构变换,实现质子从阳极到阴极源源不断的运输。该体系一般采用的电池结构如图12 所示,其中Li 源一般选择LiBF4或者LiClO4,可以采用Cu、Ag、Mo、Fe 等金属作为阴极,溶剂一般采用四氢呋喃(THF),该物质具有较高的N2溶解能力,其溶解度约为6 mmol•L-1(25℃)[14],相比水溶液体系能够有效强化N2的溶解及传质过程。另外,金属锂的独特之处在于它可以自发地在环境条件下分裂氮氮叁键,以实现N2的高效活化过程[54],基于Li介导体系的流程及特点,其有望能够在电催化NRR领域中实现新的突破。

图11 锂介导的氮还原催化系统中的物质循环[53]Fig.11 Material cycling in a lithium-mediated nitrogen reduction catalytic system[53]

图12 锂介导电催化氮还原合成氨的反应装置[54]Fig.12 Reaction setup of the lithium-mediated nitrogen reduction catalytic system[54]

Lazouski等[53]采用锂介导体系,以钢网作为阴极,溶液体系为1.0 mol•L-1LiBF4、0.1 mol•L-1EtOH(THF),实现了30 nmol•s-1•cm-2的产氨速率以及35%±6%的法拉第效率。Andersen等[55]也在该体系中以Mo片作为阴极,电解质溶液为0.3 mol•L-1LiClO4、1%(体积分数)EtOH(THF),实现的产氨速率为0.7 nmol•s-1•cm-2,法拉第效率为37%。虽然Li 介导体系已有许多报道,但性能并不是特别突出,阻碍该体系发展的主要原因为乙醇作为质子载体其在催化过程中不能完全实现循环,其在阳极可能被氧化从而不能够继续作为质子载体,因而现有研究中乙醇普遍充当牺牲试剂,其在电化学过程中不断被消耗。
基于上述乙醇在Li介导NRR 体系中的特点,实际的运行并不是可持续的连续过程,并且乙醇不断被消耗会对该体系电催化的性能造成影响。针对这一问题Suryanto 等[56]对质子载体进行研究,将乙醇替换为[P6,6,6,14]+,如图13所示,实验证明,[P6,6,6,14]+具有更高的化学稳定性、电化学稳定性以及热稳定性,其能够实现可逆的脱质子过程,从而在阴、阳两极间可持续地进行质子运输,同时[P6,6,6,14]+体系对于N2具有较高的溶解能力,因而其更适合作为Li介导体系的质子载体。采用该质子载体,研究者实现了(53±1)nmol•s-1•cm-2的产氨速率以及69%±1%的法拉第效率,并且该体系能够可持续地连续运行3 d以上,具有一定的催化稳定性,性能十分优越,说明此种改性策略的有效性,该研究使得电催化NRR的性能前进了一大步。

图13 从H2和N2可持续性电催化合成氨流程示意图[56]Fig.13 Schematic illustration of sustainable electrosynthesis of ammonia from H2and N2[56]
除Li介导体系外,Al-N2非水体系也取得一定的研究进展,Guo等[57]以铝作为阳极,以石墨烯负载钯作为阴极,电解质选用氯铝酸型离子液体构建了Al-N2电池体系,如图14所示,电池放电过程中,阳极铝溶解为Al2Cl-7进入电解质中,而后迁移至阴极与N2反应生成AlN和AlCl-4,其产物AlN可在碱性溶液中转化为NH3·H2O从而实现氨的合成。该体系中,离子液体的选用有效抑制了析氢反应的发生,同时从Gibbs自由能角度分析AlN比LiN3更容易形成,因而对N2具有较高的活化能力,研究实现了27.1 mg•(g cat)-1•h-1的氨收率以及51.2%的法拉第效率。

图14 Al-N2电池系统中能量存储及N2固定过程实现示意图[57]Fig.14 Schematic diagram of energy storage and N2 fixation process in the Al-N2 battery system[57]
在非水体系的研究中,常采用非质子溶剂作为电解液,能够有效抑制析氢反应的发生,同时,通过合理设计构建装置中的电化学过程也能够实现原料N2的高效活化,因而相关研究展现出较高的电催化NRR 活性及选择性,非水体系的构建及优化也是本领域中电催化性能提升的重要突破口之一。
3.5 电解池的工作电极电位
电催化NRR 过程进行的重要工作条件之一即为电池电压,特别是阴极的工作电位能够直接影响氮还原反应的速率及选择性,因而工作电压的选择也是十分重要的。Chen 等[38]采用Fe2O3-CNT 作为催化剂、0.5 mol•L-1KOH 作为电解质,考察了不同工作电压下(-1.0、-1.5、-2.0 V)的电流密度及产氨量,如图15 所示,随着电池电压的增加,电极反应的动力学加快,电流密度增加,产氨速率增加,对应于同一反应时间,工作电压大的体系产氨量更高。
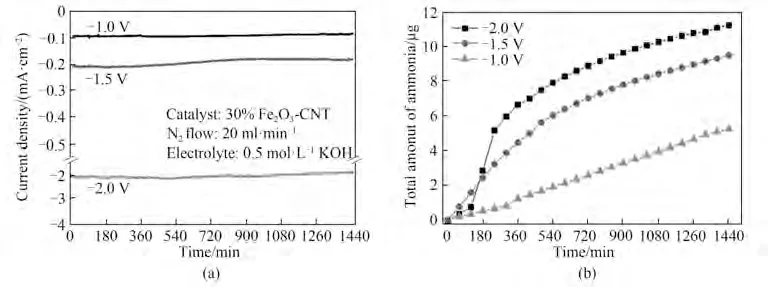
图15 工作电极电位(vs Ag/AgCl)对电流密度及总产氨量的影响[38]Fig.15 Effect of working electrode potential(vs Ag/AgCl)on current density and total ammonia production[38]
但同时研究者发现工作电压的加大会造成法拉第效率的减小,Cheng等[58]以Fe3Mo3C/C作为催化剂,通过不断降低阴极电位,如图16 所示,产氨速率先升后降,于-0.5 V 处取得峰值,而法拉第效率随着电极电位的负移而逐渐降低,亦即在一定范围内,电位负移促使产氨速率增加是以牺牲法拉第效率为代价的。阴极电位负移,电池电压加大,反应动力学加快,产氨速率增加,然而氮还原反应与析氢反应的电位十分接近,且动力学上H+比N2更加容易活化,产氨速率加快的同时,析氢反应速率也有显著提高,甚至提升程度比NRR 更大,造成法拉第效率的降低。因而一味加大电池电压并不是提升电催化NRR 性能的有效措施,实际工况中,工作电压的选择需要在产氨速率与法拉第效率二者间进行权衡。

图16 Fe3Mo3C/C在不同电极电位下的产氨速率及法拉第效率[58]Fig.16 Ammonia production rate and Faradaic efficiency of Fe3Mo3C/C at different electrode potentials[58]
3.6 其他优化策略
3.6.1 阳极反应的替换 常规电催化NRR 体系中阳极上发生的是析氧反应(OER),由于OER 动力学缓慢影响整体的电解速率,致使需要很大的过电位来驱动反应,OER半反应决定体系整体的电池电压,且产物氧气的附加值不高,因而有研究试图将阳极反应替换为电极电位更低的反应。Zhao等[59]将葡萄糖酸钠引入电解质溶液中,用氧化葡萄糖酸钠(SG)为葡糖二酸(GA)的反应替换原阳极的OER,不影响NRR 活性的前提下,将体系所需电池电压降到0.4 V。类似地,还可以将阳极OER替换成甘油氧化为甘油醛[60]、5-羟甲基糠醛(HMF)氧化为2,5-呋喃二甲酸(FDCA)[61]等反应,能够减小体系电池电压的同时联产高附加值产品,提升工艺过程的经济性。
3.6.2 加压体系的构建 由于NRR 与HER 的电极电位十分接近,且质子在动力学上比N2更容易活化,因而选择性抑制析氢反应的难度较大,有研究创新性地从改变化学平衡的角度对此问题进行解决,Cheng 等[58]注意到,如式(8)和式(9)所示,由于氨在水中的溶解度极高,氮还原合成氨电极反应的正向反应过程气体体积减小;相反,HER 的正向反应气体体积增加,升高压力可以同时实现NRR 的促进与HER 的抑制。如图17 所示,该研究将常温常压下的电化学装置转移至压力釜中进行加压操作,通过实验验证了加压对于析氢反应的有效抑制作用,并在0.7 MPa 的条件下实现了14.74%的法拉第效率。另外,研究发现升高压力能够强化N2的溶解和传质过程,从而提升相同条件下的电流密度,同时加压能够显著降低电池所需的工作电压,在更小的工作电压下实现更高的法拉第效率[58]。

图17 不同加压条件下Fe3Mo3C/C的产氨速率及法拉第效率[58]Fig.17 Ammonia production rate and Faradaic efficiency of Fe3Mo3C/C under different pressure conditions[58]
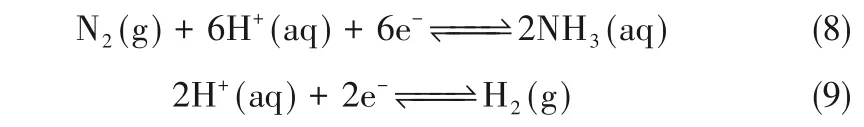
3.6.3 氨浓度准确检测流程的优化 现有研究中氨含量的检测方法主要有:纳氏试剂分光光度法、靛酚蓝比色法、氢谱核磁法、离子色谱法等[62-66],其中前两种方法更为常用。由于大多数文献报道中电催化氨合成的产率并不高,反应结束后电解液中的氨浓度较低,因而氨浓度的检测结果很容易受到原料气、电解槽、质子交换膜、电解液、电极、催化剂、玻璃器皿、实验用耗材,甚至人的呼吸气中含氮杂质的影响,从而造成假阳性的结果[67]。为避免引入此类误差,一方面,在对电极体系中电解槽、电解液、催化剂等的选择过程中,尽量避免使用含氮材料。同时规范实验操作,避免在实验过程中引入含氮杂质。另一方面,如果装置体系有含杂质的可能,则需要采取必要的纯化措施[68]。如对于原料气的纯化,可采用硫酸作为吸附剂除去原料气中的NH3杂质;亚硝酸盐、硝酸盐、二氧化氮等可溶性含氮杂质可通过水洗的方式除去;对于一些水溶性较差的氮氧化物,可以采用选择性催化还原法进行除杂而后再经过硫酸洗涤除去多余的NH3[69],或者采用双氧化剂溶液(H2O2/S2O2-8)在50℃、pH=11 的条件下将其氧化为N等可溶性杂质后再进行除杂[70]。若电解质溶液或者电极存在含氮杂质,则可经过高温处理或KOH 溶液洗涤等方式进行除杂;对于质子交换膜,如Nafion膜,除需经过H2O2、H2SO4溶液浸泡等必要的除杂及活化过程外,还可进一步将预处理好的膜在超纯水和稀硫酸溶液中进行超声处理,以消除混在膜中的含氮杂质的影响。采取有效措施对电化学体系的含氮杂质进行消除的同时,还需要注意避免在除杂过程中产生新的污染物。
为保证氨含量测试结果的准确性,一般还需进行同位素实验或空白实验排除含氮杂质的影响。Andersen 等[71]报道了同位素实验定量测量氨产量的方法,该方法准确度和灵敏度较高,能够实现对于氨浓度低至1 μmol•L-1体系的检测,检测过程无须将氨从电解液中分离,并且能够通过此法证明合成的氨来自原料氮气的转化,是目前本领域研究中合成氨浓度的有效测量手段。基于上述研究报道,本领域研究者在进行相关实验过程中有必要根据具体体系采取有效的除杂措施,并通过同位素实验或空白实验进行定量分析,同时证明合成的氨来源于原料氮气,避免假阳性结果的产生,保证结果的有效性。
4 总结及展望
综上所述,电催化氮还原合成氨工艺路线近几年已成为研究热点,尽管目前该工艺所实现的产氨速率以及法拉第效率还不能够满足商用化工业生产,但随着对机理、催化材料、装置体系的不断深入研究,该领域的成果日益丰富,并且性能上也取得了显著进步。未来的发展仍需针对该领域中的关键技术难点,从关键科学问题出发,围绕催化材料和装置体系两大方面继续深入研究。其中关于装置体系未来可以继续从以下几方面进行突破。
(1)优化反应器结构。电催化反应器整体结构为工艺的运行提供稳定的工作环境,其结构形式直接决定了阴、阳极室的连接方式,电极的排布等,现有研究中采用的反应器形式有背对背型、聚合物电解质膜型、H型以及单槽型四种,未来研究可以根据具体的体系,选择合适的反应器形式,并对其结构参数进行优化。另外,研究可以针对电催化NRR 体系的需求进行新型反应器的设计与开发,例如可以将微反应器与电池结构的设计进行结合,利用微反应器极小的特征尺寸,实现电催化反应中传质等过程的强化。
(2)优化电极结构。电极是电化学反应发生的主要场所,由于电催化NRR 的工作电极一般是将催化剂负载于集流体上,因而对于电极的优化大部分工作是对催化材料的设计及改性,在高活性催化材料构建的基础上,可以对电极本身结构进行优化,例如构建多层电极结构、引入非质子疏水保护层、引入“金属绝缘体-催化剂”结构等改进电极上质子、电子的传递途径及速率,控制催化材料附近的质子、电子浓度,从而抑制析氢反应,提升反应选择性。未来需要进一步设计阴极的电极结构,使其质子、电子传递途径更为合理,为电极反应的进行创造良好的条件。
(3)优化电解液组成。电解液直接决定了电极所处的溶液环境,其中发生离子运输、反应原料扩散等重要过程,现有研究中多采用水溶液作为电解液,因而可以对水溶液的组成及配比进行优化,一方面增强电解液对N2的溶解能力,增强传质过程;另一方面调控电解液中质子的传递方式及速率,从而对析氢反应进行抑制。未来研究可以根据分子结构以及电荷相互作用等,继续探索N2溶解能力高、质子传递速率适中的水溶液体系。
除此之外,还可构建非水体系作为电解液,如离子液体等。非水体系中Li 介导的反应体系的性能更为突出,其采用N2溶解能力较强的THF 作为溶剂,通过载体完成质子在两极间的运输,近期刊登在Science上的研究成果采用稳定性更高的磷盐作为质子载体,实现了Li介导体系电催化NRR性能的新突破,未来Li 介导体系的开发必然成为该领域中研究热点。
(4)优化反应的操作条件。在反应器形式、电极结构、电解液组成确定后,实际电催化过程的运行还需选择合适的操作条件,因而需要对体系的温度、压力、工作电位等参数的影响规律进行研究,探索合适的操作条件,从而保证电催化NRR 时刻处于最优的运行状态,提升工艺的经济性。
尽管优化的策略种类丰富,但归根结底都是以克服工艺的关键技术难点为核心目标,即提升体系对N2的活化能力、有效抑制阴极上析氢反应、强化N2的溶解及传质过程,从而实现工艺在活性及选择性方面的突破。目前电催化NRR 工艺虽然没有Haber-Bosch 工艺成熟,产氨速率、法拉第效率等性能参数仍有待提高,但随着研究的进行,该工艺的性能已实现了显著的提高,并且该领域的研究热度正逐步上升,未来需继续围绕催化材料、电催化装置体系两个方面进行深入探索,早日实现电催化NRR工艺的工业化应用。
