石墨烯基PtCu二元金属催化剂对甲醇吸附性能的理论研究
2021-12-27佟永纯王春艳孙启稿王清云
佟永纯, 张 丹, 王春艳, 孙启稿, 王 欣, 王清云
(河西学院 化学化工学院 甘肃省河西走廊特色资源利用重点实验室, 张掖 734000)
1 引 言
燃料电池(Fuel Cell)是一种将化学能等温、直接地转化为电能的发电装置,被广泛的应用于军事、航天、机动车、移动设备、居民家庭等领域,具有很好的应用前景. 其中,燃料电池中的直接甲醇燃料电池(DMFC)具有比能量高、清洁、使用安全方便、结构简单等优点,在便携式电源和车载电源等领域尤为突出. 目前燃料电池的性能与商业化的要求仍有较大的差距,引起了人们的广泛关注[1].
但是,甲醇氧化过程(MOR)涉及六电子转移,反应过程复杂、过电位高、动力学速率缓慢,因此研究和开发高活性、高稳定性的MOR电催化剂一直是DMFC研究的热点和难点. 目前直接甲醇燃料电池使用最普遍的催化剂是Pt基催化剂,然而由于贵金属Pt催化剂存在资源稀少、价格昂贵、纯Pt催化剂易中毒、利用率低等问题,阻碍了直接甲醇燃料电池的商业化进程. 因此提高提高贵金属的利用率或寻找新的替代贵金属的催化剂是推进直接甲醇燃料电池商业化进程的有效途径. 故人们开始用廉价的3d金属合金化Pt,形成二元催化剂[2-10]. 这些二元金属催化剂具有与纯Pt催化剂相似的催化活性,甚至更好. 例如PtCo、PtRu、PtPd、Au、PtNi、PtMo、PtCu、PtFe、PtSn等,其中PtRu是目前性能最好的二元金属催化剂.
金属Cu由于价格低廉,并且与Pt形成的二元合金具有较好的催化性能,越来越受到人们广泛关注. Richa课题组使用浸渍还原法制造了具有不同摩尔比的PtCu催化剂,并采用循环伏计量、电化学阻抗光谱和电子转移测量中发现,合成PtCu(1:2)/N-rGO催化剂的电催化活性远远高于Pt/N-rGO和Pt/C催化剂的电催化活性[11]. 同时我们课题组研究发现PtCu催化剂具有良好的抗CO中毒性[12]. 石墨烯-PtCu复合物展现了比石墨烯-Pt和商业石墨烯-Pt更优良的电化学活性和对于甲醇催化氧化的高耐毒性,这表明石墨烯-PtCu可以成为有前景的直接催化甲醇燃料电池(DMFC)的催化剂. 石墨烯具有优异的电、力和热学等性能,因此其可作为金属原子的载体材料来实现新的催化性能. 通过研究表明,金属掺杂石墨烯结构有着非常优良的性能,过渡金属与石墨烯的相互作用使得金属掺杂石墨烯成为高活性的催化剂[13].
由于铜对环境无害,使用方便,而且地球上储量也丰富,所以近几年有很多关于铜掺杂石墨烯的研究[14-16],因此我们将在此基础上进一步研究二元金属掺杂石墨烯,即PtCu掺杂石墨烯. 在实验上很多科研工作者已经对CH3OH在PtCu/graphene上的催化性能进行了一定的研究,但是理论上的研究上的研究比较少,尤其是对其微观掺杂模型的研究. 因此,在本文中我们将通过密度泛函理论研究CH3OH在PtCu/graphene的吸附机制及PtCu二元金属催化剂在石墨烯上的微观吸附模型.
2 计算方法
本文均采用密度泛函(DFT)计算,以Dmo13模块来执行[17]. 广义梯度近似 (GGA)的Perdew-Burke-Ernzerhof(PBE)函数用来描述交换一关联效应[18],并用Mulliken电荷分析方法[19],对涉及到的电子转移情况进行了分析. 计算时我们选取了p极化函数的双数字基组(DNP)[20]对该论文所研究体系进行了梯状密度泛函计算,该计算已成功地应用于金属-半导体表面的相互作用的研究. 通过用一个Pt原子取代含有32个C原子的4×4的超晶胞石墨烯中的其中一个C原子来计算研究. 为了使石墨烯之间尽量没有相互作用,我们把z方向上的晶格矢量设为2 nm. 在结构优化时,把布里渊区内k值设为3×3×1,这使得能量收敛误差为2.7×10-4eV,力的最大收敛标准为0.54 eV/nm. 对于CH3OH在石墨烯上的吸附能通过以下的公式来定义:
Eads=Etotal-(E1+E2)
其中E1为Pt、Cu、PtCu掺杂在石墨烯载体上的总能量,E2为CH3OH吸附的总能量,Etotal为甲醇吸附后的总能量. 同时吸附能越小,意味着效应的结构体系越稳定.
3 结果与讨论
3.1 Pt、Cu、PtCu掺杂在石墨烯上的几何构型
Pt、Cu掺杂在石墨烯上时,Pt或Cu取代了石墨烯上的一个C原子,金属Pt或Cu与石墨烯上缺陷处的三个C成键,由于金属的原子半径比C原子半径(0.07 nm)大,所以金属原子与C原子成键后就会在石墨烯表面凸出以获得更大的空间,结构如图1所示. 对于结构L1,Pt与石墨烯缺陷处的3个C原子成键所形成的Pt-C键长分别为0.197 nm、0.197 nm、0.197 nm. 结构L2中,Cu与石墨烯缺陷处的3个C原子成键所形成的Cu-C键长分别为0.186 nm、0.186 nm、0.186 nm(见表2). 通过对比发现Pt在石墨烯上的吸附能低于Cu在石墨烯上的吸附能( L1:在GGA/PBE条件下Eads=-7.22 eV;L2:在GGA/PBE条件下Eads=-3.86 eV). 根据Mulliken电荷分析可知,石墨烯向金属Pt转移了0.170 e,向金属Cu转移了0.132 e,使得金属Pt或Cu都带有一定的负电荷(见表1).
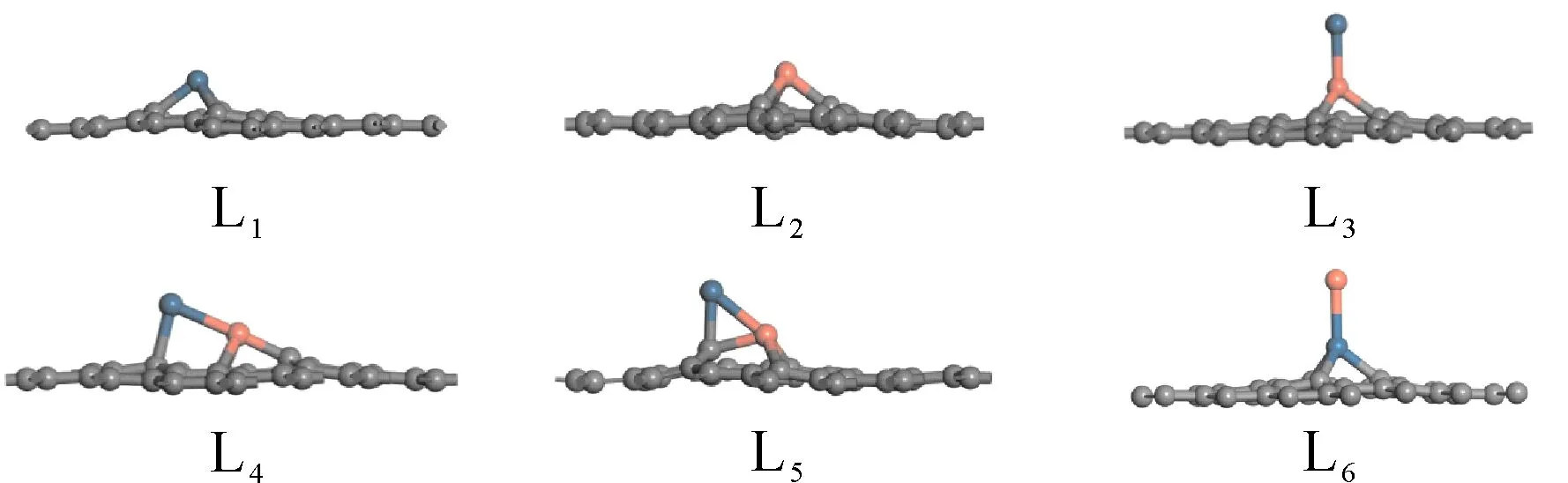
图1 Pt/graphene、Cu/graphene、PtCu/graphene结构示意图Fig. 1 The geometries of Pt/graphene, Cu/grapheneand PtCu/graphene
当Pt和Cu同时掺杂在石墨烯上时,找到如图1所示的四种可能结构. 对于L3,Pt、Cu仍然以直线形式垂直于石墨烯,但是Cu与石墨烯缺陷处的3个C原子成键,其中Pt和Cu的距离为0.228 nm,Cu和3个C原子的距离约为0.19 nm.根据Mulliken电荷分析知,Pt和Cu向石墨烯转移了0.460 e,其中Cu向石墨烯转移了0.201 e,Pt向石墨烯转移了0.186 e. L4和L5的结构明显不同于结构L3. 对于结构L4,Cu与石墨烯缺陷处的3个C原子成键,Cu不仅与相邻的C原子成键而且与Pt也有成键现象,形成了C-Cu-Pt桥,Pt-C键长为0.213 nm,3个Cu-C键长分别为0.194 nm、0.194 nm和0.188 nm,Pt与Cu的距离为0.242 nm,Pt向石墨烯转移了0.186 e而Cu向石墨烯转移了0.211 e. 对于结构L5,Cu原子与石墨烯缺陷处的3个C原子成键,同时Pt与石墨烯上临近的一个C原子成键. 其中Pt和C距离为0.198 nm,3个Cu-C键长分别0.186 nm、0.186 nm和0.196 nm,Pt-Cu距离为0.235 nm. 由Mulliken电荷分析知,Cu原子向石墨烯转移了0.203 e而 Pt 原子向石墨烯转移了0.219 e. 对于L6,其结构与L3非常相似,Pt和Cu仍然以直线形式垂直于石墨烯,但是Pt与石墨烯缺陷处的3个C原子成键,其中Pt和Cu的距离为2.431 Å,Pt和3个C原子的距离约为2.0 Å,Pt到石墨烯表面的距离比结构L1中Cu到石墨烯的距离略长. 根据Mulliken电荷分析知,Pt和Cu向石墨烯转移了0.318 e的电子.
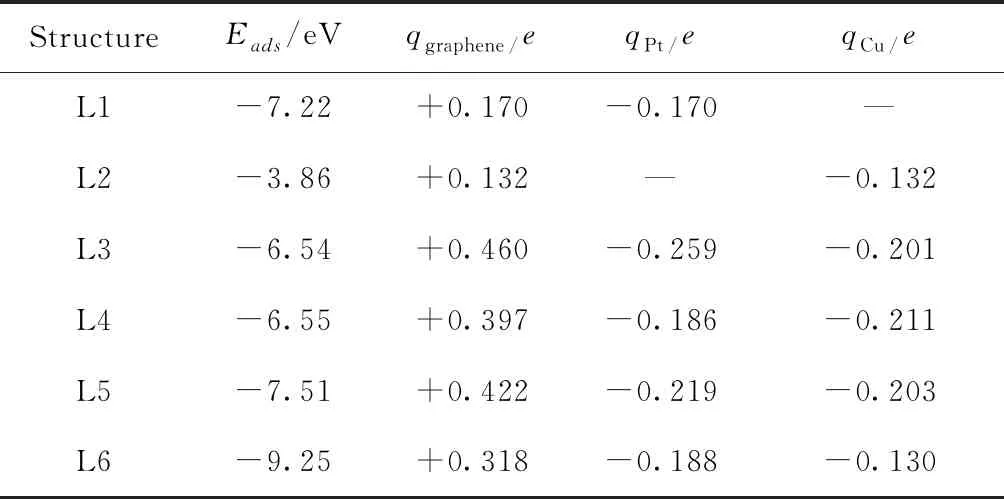
表1 Pt、Cu、PtCu吸附在单空位石墨烯上的计算参数
通过对以上四种PtCu二元金属结构的稳定性比较,L3和L4相差不大,但是对于L6的能量明显低于其它三种结构,对应的吸附能为-9.25 eV,因此L6是这四个结构中最稳定的.
3.2 甲醇在Pt、Cu、PtCu掺杂石墨烯上的吸附构型
甲醇分子在吸附过程中有羟基上的氧原子吸附(O-吸附)和甲基上的氢原子吸附(H-吸附)两种方式. 所有的结构绘制于图2,相应的计算参数列于表2和表3.
3.2.1O-吸附
O-吸附后可得L3-O、L4-O1、L4-O2、L5-O1、L5-O2、L6-O(如图2)六种结构. 在L3-O中,由于甲醇中O的吸附导致Pt-Cu与石墨烯的之间的角度有由原来的近90度变为近60度,Pt与石墨烯上C的距离也由原来的0.190 nm变为0.195 nm,对应的Pt与O的距离为0.221 nm,Cu与3个C原子的距离比L3中的Cu-C距离有拉长现象. 在L4-O1中,甲醇中的氧吸附在Pt位点上,对应的Pt-O键长为0.218 nm,同时Cu和Pt与临近C之间的距离相较于L4中金属Pt、Cu与C的距离变化不大. 当甲醇中的O吸附在Cu位点形成L4-O2时,Cu与O的距离为0.206 nm,金属Pt、Cu与石墨烯中临近C原子的距离与L4中相应的距离微有拉长. O-吸附在L5中的Pt位点和Cu位点分别形成L5-O1和L5-O2,对应的Pt-O和Cu-O键分别为0.220 nm和0.215 nm,但是对于L5-O2,由于甲醇的吸附使金属Pt、Cu与石墨烯的吸附能力明显由弱化现象(表2). 对于L6上的O-吸附,虽然L6结构是这四种二元金属催化剂中最稳定的,但是对甲醇的吸附能力确是最弱的,这说明Cu位点催化活性不是很好.
通过计算可知,甲醇中的O-吸附在Cu位点时能够降低Cu与石墨烯之间的相互作用,但是当时当O-吸附在Pt位点时却能够一定程度的增强Cu与石墨烯载体之间的相互作用. 根据Mulliken电荷分析可知(表2),当甲醇吸附在PtCu/graphene上时,甲醇中的电子流向了催化剂. 甲醇的氧端更容易吸附在L3上,对应的吸附能为-1.96 eV. 通过对以上七种结构的综合对比,发现甲醇的氧端更容易吸附在Pt上,而且在Cu的掺杂下,Pt位点的活性有所提高.
3.2.2H-吸附
对于H-吸附吸附结构,通过计算可以得到L3-H、L4-H1、L4-H2、L5-H1、L5-H2、L6-H(如图2). 在L3-H结构中,羟基中的H与Pt有成键现象,Pt-H键长为0.220 nm. 同时金属Cu和3个C原子的距离与L3几乎没有变化. 在L4-H1和L4-H2中,对应的Pt-H和Cu-H键长为0.215 nm,0.287 nm,Cu和3个C原子的距离与L4中金属Pt、Cu与C的距离略有不同. 在L5-H1中,的Pt-H键长为0.188 nm比L5-H2中的Cu与H短0.099 nm,Cu和3个C原子的距离与L5中金属Pt、Cu与C的距离比较相近(表2). 对于L6,甲醇的甲基氢更难吸附,对应的吸附能只有-0.08 eV.
根据Mulliken电荷分析可知(表1),当甲醇吸附在L3、L4、L5和L6上时,甲醇中的电子逐渐向催化剂转移,但L4-H1、L5-H1中甲醇中的电子更多的流向了载体石墨烯,对应的吸附能分别为-0.24 eV、-0.48 eV,而L3-H、L4-H2、L5-H2和L6-H的吸附能则小的多,可以看出H-吸附比O-吸附要困难. 通过在Pt中掺杂Cu对于H-吸附的吸附能力影响不大或略有增加,而对于O-吸附,Cu的掺杂能够大大增加甲醇的吸附能力.
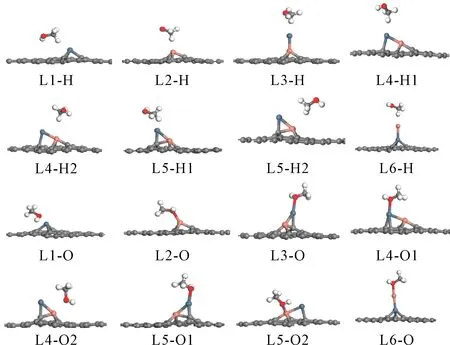
图2 CH3OH在Pt/graphene、Cu/graphene及PtCu/graphene二元金属催化剂上的吸附结构示意图Fig. 2 The geometries of CH3OH adsorption on the Pt/graphene, Cu/graphene and PtCu/graphene
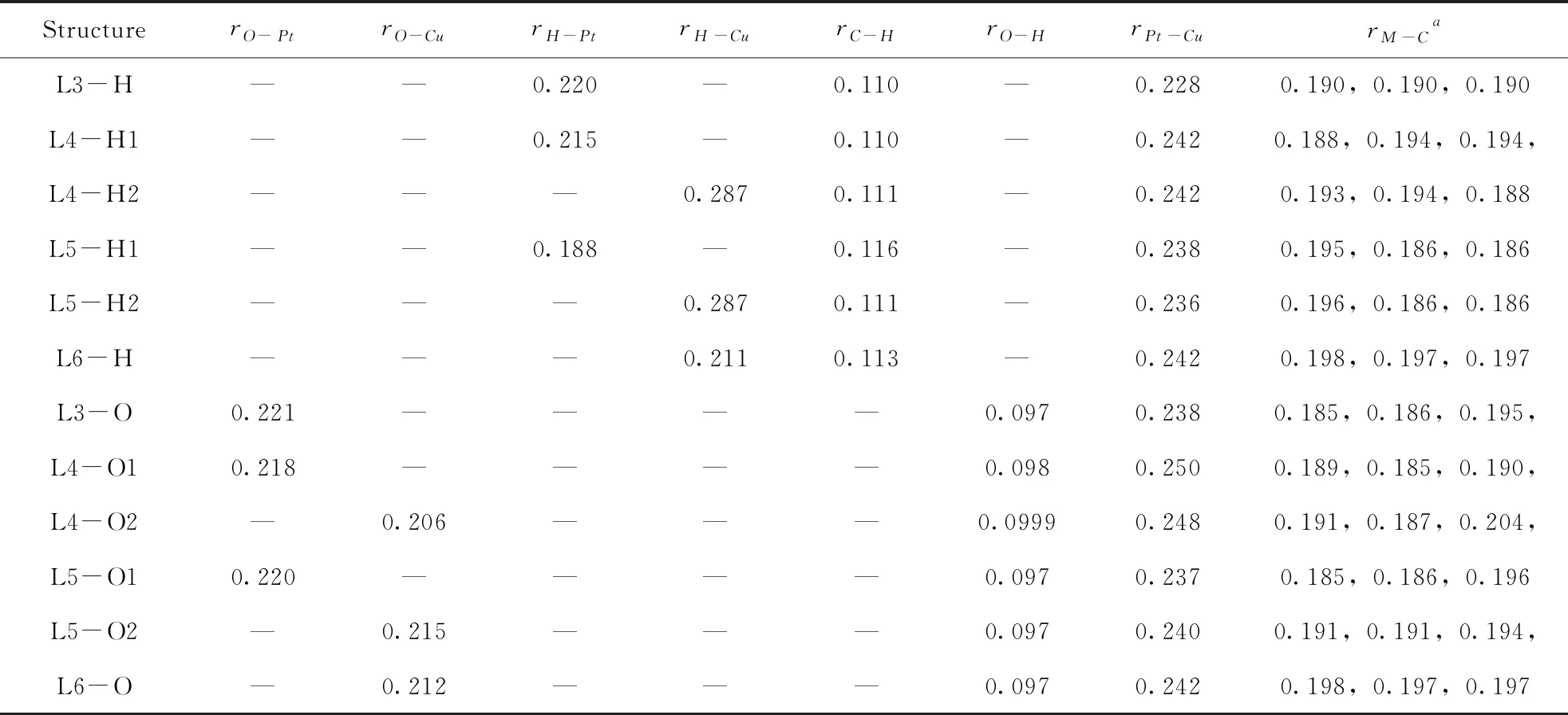
表2 CH3OH在PtCu/graphene二元金属催化剂上吸附的键长(r/nm)
3.2.3O-吸附和H-吸附的对比
对于O-吸附甲醇更容易吸附在L2上,H-吸附则更容易吸附在L1上. 对于PtCu二元金属催化剂,可以很清楚的发现甲醇更容易吸附在Pt位点上,同时对于甲醇中的H-吸附要弱于O-吸附. 为了进一步分析甲醇在二元金属催化剂上的两种吸附方式,分别计算了甲醇吸附在Pt位点上结构的HOMO-LUMO能带宽度图(见图3). 图3中浅蓝色表示L3、L4、L5的ΔEH-L;蓝色表示Pt位点上H-吸附结构中的三种稳定结构的ΔEH-L,黄色则表示Pt位点上O-吸附结构中的三种稳定结构的ΔEH-L. 一般来说,因为不利于在高激发态的LUMO中添加电子,也不利于从低能 HOMO中提取电子,所以HOMO-LUMO大的空隙与较高的动力学稳定性有关. 同时,ΔEH-L能带宽度与相对能量相似. 从图中发现,H-吸附HOMO-LUMO空隙都相对于L3、L4、L5的催化剂变化不大,而O-吸附结构HOMO-LUMO空隙都相对于L3、L4、L5的催化剂则有较大的变化,可以说明甲醇中的O-吸附使三种催化剂发生变化,使得HOMO-LUMO间隙相对较小,增加了动力学稳定性,使结构更加稳定. 在L3-O中HOMO-LUMO间隙降低最多,说明L3-O是最稳定的结构.
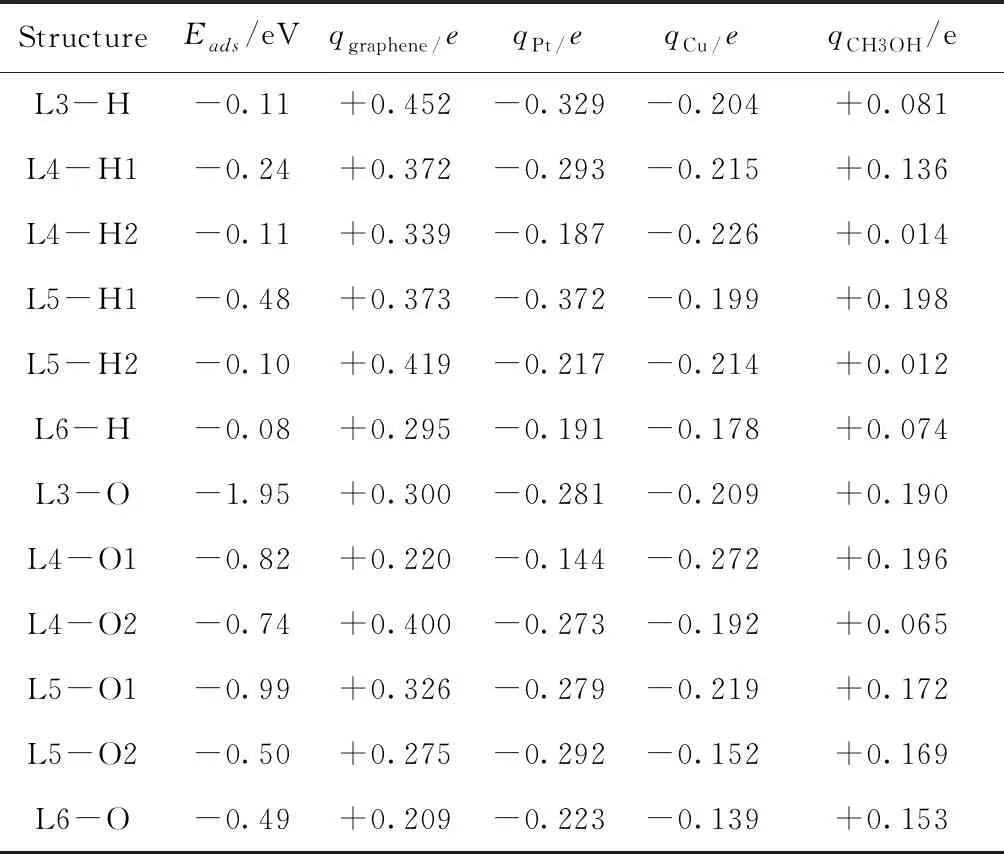
表3 CH3OH在PtCu/graphene二元金属催化剂上吸附的计算参数
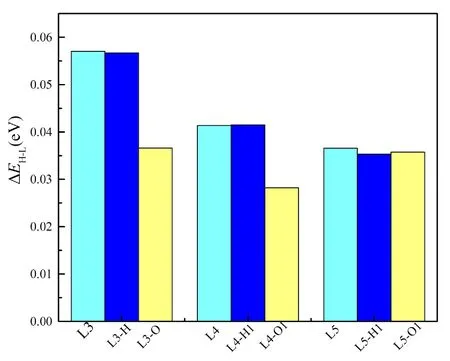
图3 甲醇吸附在Pt位点上的稳定构型的HOMO-LUMO 能带宽度图Fig. 3 The HOMO-LUMO band widths of methanol adsorption on the Pt site
4 结 论
本文通过基于密度泛函理论主要研究了PtCu的二元金属催化剂与甲醇的相互作用,通过上述分析,PtCu/graphene二元金属催化剂与甲醇相互作用中,甲醇容易吸附于Pt位点上. 对于PtCu/graphene二元金属的Cu位点的吸附能力于相对于纯Cu变化不大,但是PtCu/graphene二元金属的Pt位点相对于纯Pt,则甲醇的吸附能力却有明显的提高. 因此Cu的掺杂对于提高Pt位点的活性起到重要作用.
