用于超级电容器的木质纤维素基碳材料的制备及其性能研究
2021-12-21任晓丽刘秀知侯庆喜
任晓丽 刘秀知 刘 苇 侯庆喜,2,*
(1.天津科技大学天津市制浆造纸重点实验室,天津,300457;2.华南理工大学制浆造纸工程国家重点实验室,广东广州,510640)
随着工业的飞速发展和人口的不断增长,全球能源需求不断增加,同时伴随着人类对化石能源的过度消耗,因此造成了气候变化、能源短缺等一系列问题。因此,寻找一种高效、清洁、可持续发展的新能源及制造可实现能量储存的储能设备势在必行[1-3]。超级电容器又名电化学电容器,是一种介于传统电容器和充电电池之间的新型储能装置[4-5],广泛应用于便携式设备、电动汽车和固定储能系统中,被认为是最有前途的储能器件之一[6-7]。超级电容器中,电极是重要的储能元件[8-9]。碳材料具有加工简单、成本相对较低及良好的化学稳定性等优点,是一种良好的制备电极的候选材料[10]。木质纤维素是地球上最丰富的可再生生物质资源,来源主要包括木材、农林废弃物及其他各类能源植物[11-13]。一般情况下,木质纤维素生物质材料可从林业、农业、制浆造纸工业和木材加工行业获得[14]。木质纤维素除在上述行业或领域被利用外,最常见的处理方式是直接燃烧,该方法投资最少,但热效率低(10%~15%),不仅浪费资源,而且会造成严重的环境问题[15]。因此,将木质纤维素作为碳源制备碳材料是一种高值化利用木质纤维素的方式。
目前,常见的碳材料制备方法有模板法[16-17]、物理活化法[18]、化学活化法及细菌活化法[19]等。相比于其他几种活化方法,化学活化法具有周期短、活化温度低的优点。化学活化法常用的活性试剂有氢氧化钾(KOH)[20-21]、磷酸(H3PO4)[22-23]、氯化锌(ZnCl2)[24]等。KOH活化法由于其成熟的技术和便捷的操作而成为最常用的活化方法之一。与其他活化剂相比,由KOH活化得到的碳材料通常具有良好的微孔分布和很高的比表面积[25]。叶江林等人[26]通过KOH活化花生壳制备活性碳,其比表面积可达1790 m2/g。王程等人[27]以杏壳为原料,采用微波热裂解和KOH活化联合的方法制备碳材料,研究了活化过程中KOH用量、活化温度和活化时间对碳材料性能的影响,得到最佳条件为KOH与碳的质量比2.5∶1.0,活化温度800℃,活化时间1.5 h;所制备的碳材料比表面积为1223 m2/g。杨可等人[28]采用KOH活化法制备辣椒秸秆碳材料,研究发现:在KOH和碳的质量比3∶1、活化温度800℃、活化时间100 min条件下,碳材料的比表面积达1761.16 m2/g。然而,目前对于改变工艺制备流程从而调控碳材料的结构来提升超级电容器电化学性能的研究并不多见。
本研究选择漂白硫酸盐阔叶木浆为原料,以KOH为活化剂,采用先预碳化再活化和先活化再碳化2种不同的工艺处理方法,制备了不同结构的木质纤维素基碳材料,并对不同结构的木质纤维素基碳材料的微观形貌、比表面积、孔径分布、晶体结构及表面元素等进行了表征;探究了碳材料结构与电化学性能之间的构效关系,以期为开发新型高效的超级电容器电极材料提供参考。
1 实 验
1.1 实验原料
原料为进口漂白硫酸盐阔叶木浆(LBKP)。KOH购于上海迈瑞尔化学技术有限公司,盐酸购于天津市化学试剂一厂,乙炔黑、聚偏二氟乙烯、N-甲基-2-吡咯烷酮和泡沫镍购于山西太原力之源电池材料公司。实验所用的化学药品均为分析纯。
1.2 原料处理
将购买的LBKP浆板撕成碎片,置于蒸馏水中浸泡约12 h后,使用Vally打浆机对原料疏解30 min,疏解时控制浆浓约1%。疏解后收集浆料,置于4℃的恒温条件下平衡水分24 h后,风干待用。
1.3 木质纤维素基碳材料的制备
本研究采用2种不同方法来制备木质纤维素基碳材料。
第一种方法是先预碳化再活化。具体操作如下:首先,将绝干质量3 g的LBKP置于管式炉中直接碳化,以5℃/min的升温速率升温至600℃,保温4 h,制备得活化前驱体,命名为C-600;将C-600放入10 mL KOH溶液中,常温下浸渍2 h,105℃条件下干燥,KOH与C-600绝干质量比为2∶1;最后将上述干燥后的碳与KOH固体混合物置于Ar气氛的管式炉中活化碳化,以5℃/min的升温速率,分别升温至600℃和800℃,保温2 h。以上产物均使用过量的1 mol/L的HCl溶液洗涤,再用去离子水多次冲洗至滤液pH值为中性,干燥后置于干燥器中储存,所得碳材料分别命名为P-600和P-800。
第二种方法是先活化再碳化。将LBKP直接与KOH溶液混合,KOH与LBKP绝干质量比为2∶1,常温下浸渍2 h,105℃条件下干燥。将干燥后产物放置在Ar气氛下的管式炉中,同样以5℃/min的升温速率升温至600℃,保温2 h。将以上产物使用过量的1 mol/L HCl溶液洗涤,再用去离子水多次冲洗至滤液pH值为中性,干燥后置于干燥器中储存,所得碳材料命名为T-600。
1.4 形貌和结构表征
通过扫描电子显微镜(SEM,JSMIT300LV,日本JEOL)对样品的形貌和结构进行表征。比表面积分析仪(ASIQM0000000-6,美国Quantachrome In⁃struments)在77 K(即-196℃)下测定氮气吸附-脱附等温曲线,根据所得吸附-脱附曲线,分别利用BET、DFT、t-plot方法计算得到样品的比表面积和孔径分布。采用X射线衍射仪(XRD,D8 Advance,德国Bruker-AXS)和拉曼散射光谱仪(Raman,BTC 162E-532H,必达泰克光电科技(上海)有限公司)测定样品的物理结构。X射线光电子能谱仪(XPS,ESCALAB 250Xi,美国Thermo Scientific)用于分析样品的表面元素。
1.5 电化学性能的测试
所有材料的电化学性能均是在室温下采用三电极体系(工作电极、对电极、参比电极)进行测试,所用仪器为上海辰华仪器有限公司CHI660E型电化学工作站。工作电极的制备方式:首先称取固体绝干质量比为8∶1∶1的木质纤维素基碳、乙炔黑和聚偏二氟乙烯,将聚偏二氟乙烯加入N-甲基-2-吡咯烷酮,搅拌均匀;然后将木质纤维素基碳与乙炔黑加入研钵中研磨均匀,在其中滴入加有聚偏二氟乙烯的N-甲基-2-吡咯烷酮,不断研磨使所有物质混合均匀至黏流状,再将其均匀涂敷在裁剪好的表面积2 cm2的泡沫镍上;在60℃真空环境下干燥12 h,取出后在10 MPa下压片30 s;最后进行称重,并计算涂覆的活性物质的质量。另外,分别使用铂电极和饱和甘汞电极作为对电极和参比电极,电解液为6 mol/L的KOH溶液。在-1~0 V的电压窗口内,进行循环伏安法(CV)和恒流充放电(GCD)测试,电化学阻抗谱(EIS)的测试频率范围0.01~100 kHz,交流信号振幅为5 mV。
电极的质量比电容(Cs,F/g)通过GCD计算,计算方式如式(1)所示。

式中,I为放电电流,A;Δt为放电时间,s;m为单电极片活性材料的质量,g;ΔU为电压变化范围,V。
2 结果与讨论
2.1 形态与结构分析
图1为不同结构木质纤维素基碳材料合成工艺示意图。如图1所示,采用2种不同的工艺处理方式制备得到了不同结构的木质纤维素基碳材料。图2是不同结构木质纤维素基碳材料的SEM图。从图2(a)、图2(c)和图2(d)可以看出,C-600、P-600和P-800整体结构依然保持纤维状,其局域结构表现出堆垛的褶皱状,有利于增加比表面积,从而促进离子在其表面的储存,提升超级电容器的电化学性能;然而,先活化再碳化的T-600并没有保持木质纤维素本身的纤维状结构(图2(b)),反而呈现出二维片状多孔结构。通过对比P-600、P-800和T-600的SEM图可以发现,不同的工艺处理方法可以获得不同结构的碳化材料,其结果归因于先预碳化再活化的处理工艺中,预碳化将木质纤维素表面和体相的羟基结构破坏,发生脱水反应,部分C—C转变为C=C的共轭结构[29],KOH无法破坏C=C,仅能打断体相中部分C—C,产生多孔结构;然而,先活化再碳化的处理工艺中,KOH与木质纤维素发生剥皮反应,使大部分纤维素分子链还原性末端葡萄糖基脱落,降低分子聚合度,促进KOH消除纤维素链间氢键,破坏纤维素的结晶构造,从而产生了木质纤维素的层间剥离,而且在高温碳化过程中,过量的KOH与降解产物充分反应,产生大量孔径,最终得到了二维片状多孔结构[30]。需要特别强调的是,先活化再碳化的处理方法获得的二维结构在高于600℃的条件下进行碳化,最终没有得到碳化材料,因此经过先活化再碳化处理的二维多孔结构具有高于600℃的热稳定性。
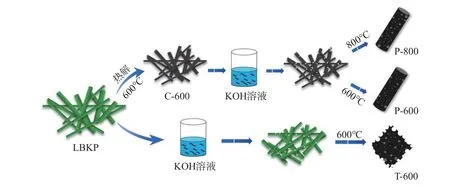
图1 不同结构木质纤维素基碳材料合成工艺示意图Fig.1 Schematic illustration of the synthesis process of lignocellulose-based carbon materials
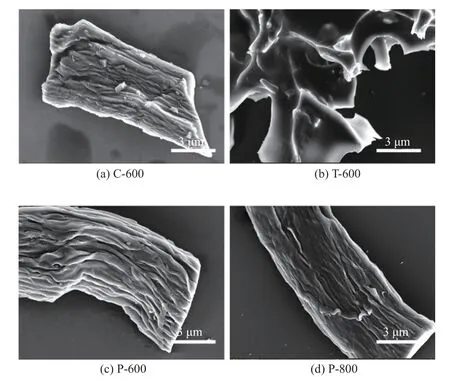
图2 不同结构木质纤维素基碳材料的SEM图Fig.2 SEM images of lignocellulose-based carbon materials
为了进一步研究不同结构木质纤维素基碳材料的表面和孔结构特征,进行了吸附-脱附等温曲线以及孔径分布的测试,结果如图3所示。根据国际纯粹与应用化学联合会(IUPAC)分类,C-600、T-600、P-600和P-800均符合I类吸附等温线(图3(a))。从图3(b)中可以看出,不同结构的木质纤维素基碳材料展现出不同的孔径分布,其中T-600、P-600和P-800均表现出明显的微孔结构,而C-600没有观察到其孔结构,这证实了上文中所提到的KOH在工艺处理过程中起到了造孔的作用;另外还可以看出,先预碳化再活化的P-800和P-600比先活化后碳化的T-600展现出更多的微孔结构,其中P-800具有更宽的孔径分布,孔径范围0.5~1.5 nm。如表1所示,采用BET计算方法得到C-600、P-600、P-800和T-600的比表面积依次为516 m2/g、1048 m2/g、1737 m2/g和954 m2/g。可以看出,无论先预碳化再活化还是先活化再碳化所获得的碳材料,其比表面积远大于直接碳化所获得C-600的比表面积,这说明KOH的造孔作用大幅度提升了碳材料的比表面积;P-800的比表面积远大于P-600和T-600,可能是由于在800℃的碳化过程中产生了更多的微孔,所获得的不同结构木质纤维素基碳材料的孔体积数据证实了这一推论。总体来说,不同结构的木质纤维素基碳材料比表面积和孔径的差异主要归因于KOH的造孔作用、碳化温度以及工艺处理步骤,其中P-800的高比表面积和丰富的孔径归咎于其自身的多孔结构和空间堆垛褶皱。

图3 吸附-脱附等温曲线以及孔径分布图Fig.3 Adsorption/desorption isotherms curves and pore size distributions

表1 样品比表面积和孔特征Table 1 Specific surface area and pore characteristics of the test samples
为深入分析不同结构木质纤维素基碳材料的晶体结构、缺陷程度以及化学组成结构,对其进行了XRD、Raman光谱和XPS表征。图4(a)为不同结构木质纤维素基碳材料的XRD曲线。如图4(a)所示,29°附近的宽峰和43°附近的弱宽峰分别归属为无定型石墨的(002)和(100)衍射峰,说明不同结构的木质纤维素基碳材料中均存在一定量的碳的本征缺陷,该本征缺陷有利于暴露更多的表面储钾位点,从而提升超级电容器的电化学性能。图4(b)为木质纤维素基碳材料的Raman光谱图,从图中可以看出有2个明显的峰,1350 cm-1和1580 cm-1的峰分别归属为D峰和G峰,其中D峰代表了碳原子晶体的缺陷,G峰代表了碳原子sp2杂化的面内伸缩振动。通常D峰和G峰的强度比(ID/IG)用于评价碳材料的缺陷程度[31];经过计算,P-800、P-600、T-600和C-600的ID/IG依次为0.96、0.77、0.85、0.65,说明P-800中存在着更多的缺陷,这与XRD分析结果一致。为了确定缺陷的类型,对P-600和P-800进行了XPS表征,结果如图4(c)及图4(d)所示。从图4(c)可以看出,P-600和P-800在284 eV和532 eV处的峰分别对应于C 1s和O 1s,这说明所获得的碳材料中仅含有C和O元素。进一步对P-800和P-600的C 1s进行分峰拟合得到3个不同的峰,分别位于284.6 eV、286.5 eV和288.7 eV附近,根据结合能的大小分别归属为C=C/C—C、C—O和C=O。从图4(d)中可以看出,P-800碳的本征缺陷所占的比例小于P-600所占的比例,而C—O/C=O在P-800和P-600中所占的比例与其正好相反,说明在P-800中存在更多的含氧缺陷位点,相比于碳的本征缺陷具有更强的储钾能力[32]。
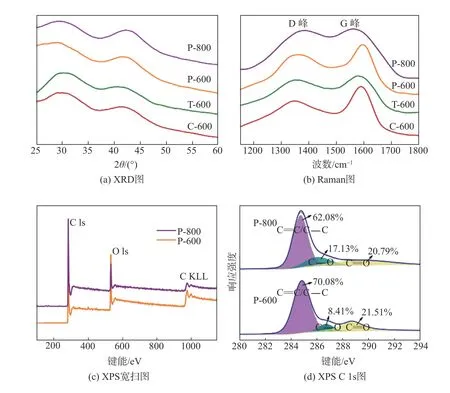
图4 木质纤维素基碳材料的结构分析Fig.4 Structural analysis of lignocellulose-based carbon materials
结合以上对不同木质纤维素基碳材料的结构分析,可以预测P-800的比表面积、孔结构和丰富的缺陷为超级电容器提供了更加优越的储钾位点。
2.2 电化学性能
为了证实上述的构效关系,对不同结构的木质纤维素基碳材料采用三电极体系,以6 mol/L KOH水溶液为电解质,电压窗口为-1~0 V进行了电化学性能的测试。图5(a)为0.5 A/g电流密度下的GCD曲线图。由图5(a)可见,所有样品在充放电过程中,均呈现出准对称三角形的相对线性的电压-时间关系。这表明所有样品作为电极均形成双电层电容器,具有显著的可逆性,所有样品的容量主要由电极和电解液之间的电子转移决定。木质纤维素基碳材料的质量比容量依据GCD曲线通过式(1)计算,在0.5 A/g电流密度下,P-800、P-600、T-600、C-600的比电容依次为194 F/g、140 F/g、111 F/g、47 F/g。其中,P-800的比容量最大,这归因于P-800相比于其他木质纤维素基碳材料具有更大的比表面积,从而提供了更多的储钾位点,多孔分布有利于K+的传输,缺陷特别是含氧缺陷位更有利于电荷的快速转移[33]。
图5(b)为不同电流密度下P-800样品的GCD曲线。0.5 A/g、1 A/g、2 A/g、5 A/g、10 A/g电流密度下的GCD曲线通过式(1)计算得到,P-800的质量比电容依次为194 F/g、176 F/g、160 F/g、122 F/g、79 F/g。在不同电流负载下呈高度线性对称三角形,进一步证实了其优良的电容性能。随着充放电电流密度的增大,质量比电容呈现降低的趋势,这可能是由于电流密度越高,电荷转移速度越快,部分电荷无法直接接触到电极表面完成吸附。图5(c)为木质纤维素基碳材料的循环伏安(CV)曲线。CV曲线积分面积是衡量电极材料性能的一个指标。从图5(c)中可见,所有样品的CV曲线都显示出电化学电容器所特有的准矩形形状。其中,P-800曲线的积分面积远大于其它样品,此结果与GCD曲线所得结果相一致。
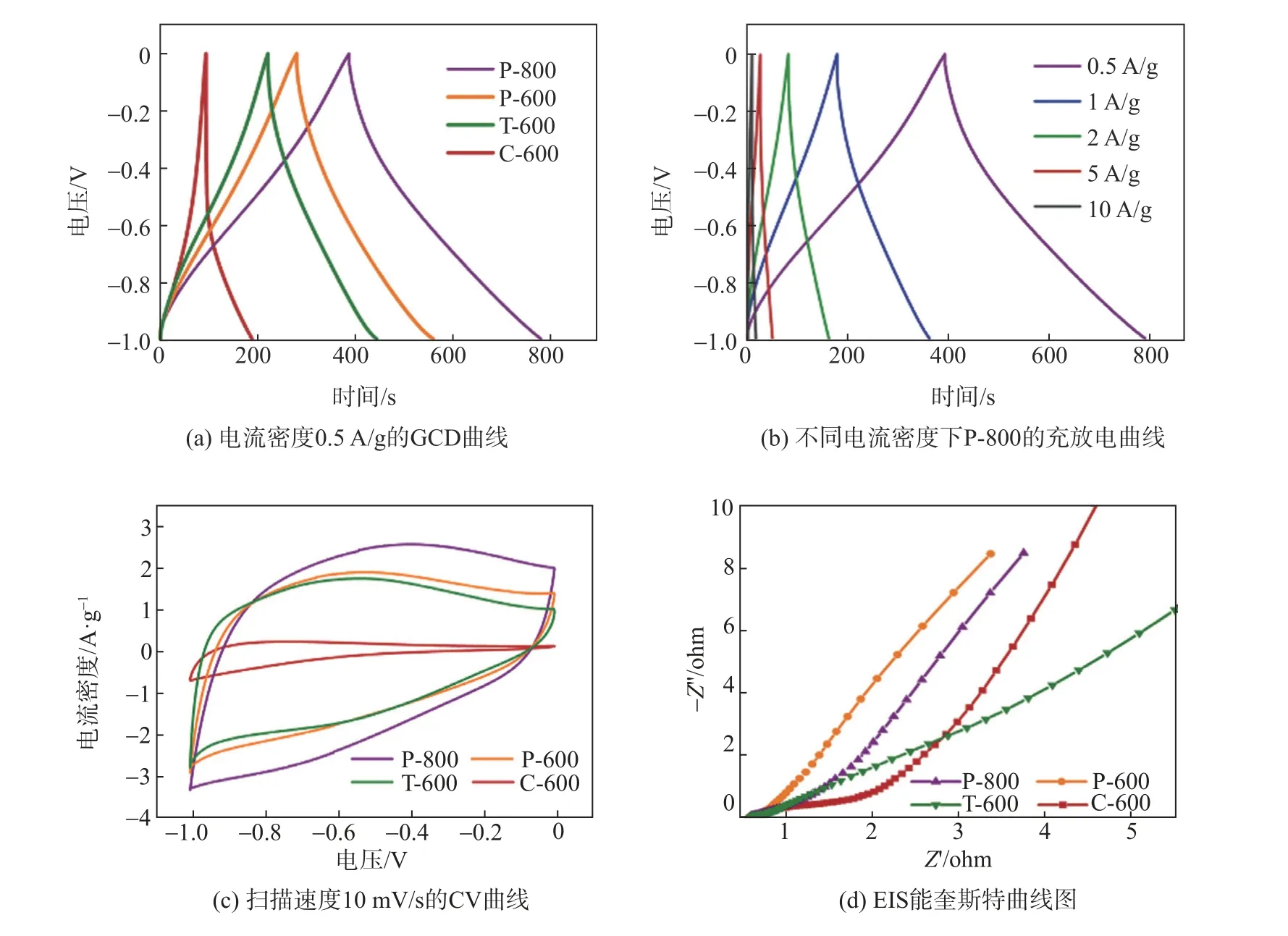
图5 样品电化学性能Fig.5 Electrochemical performances of samples
电化学交流阻抗通常用来研究电极在电荷储存过程中的电容行为。样品在6 mol/L KOH电解液中的交流阻抗(EIS)能奎斯特曲线图如图5(d)所示。在低频区,与C-600和T-600相比,P-600和P-800作为活性物质制备的电极,其交流阻抗曲线斜率更大,这意味着其传质阻力相对较低。这一结果表明,大的比表面积更有助于电解质离子的低阻扩散。高频区分为2个重要的部分:一个是小半圆的半径,表示电解质与电极之间的电荷转移电阻;另外一个是Z'轴上的截距,表示等效串联电阻,包括材料内阻、界面接触电阻、电解液电阻[34]。从图5(d)发现,P-800、P-600和T-600的半圆半径均明显小于C-600。由此可见,多孔结构有利于K+的扩散,丰富的缺陷位点降低了电荷转移阻力。P-600和P-800的瓦氏效应线更短,表明了更大的比表面积和丰富的孔径分布,有利于电荷在电极内部的快速扩散。对比截距可得到,P-600和P-800的导电性能更好,这可能是由于木质纤维素基碳材料中缺陷增加了电子的传导速率[35]。另外,P-800的截距略微小于P-600,这可能是因为P-800的含氧缺陷位相对较多,改善了碳材料的润湿性,增大了电解质与材料的相界面,从而降低界面电荷转移阻力[36]。
上述结果表明,经过先碳化再活化得到的P-800具有最佳的电化学性能,其原因为P-800自身的高比表面积、多孔结构以及丰富的缺陷位点共同提升了表面储钾能力。因此,构筑高比表面积、多孔结构和丰富的缺陷位点的木质纤维素基碳材料可以为开发新型高效的超级电容器碳材料开辟新的途径。
3 结 论
本研究以漂白硫酸盐阔叶木浆为原料,KOH为活化剂,采用先预碳化再活化和先活化再碳化2种方法改性制备了用于超级电容器电极材料木质纤维素基碳材料。结果表明:在不同结构的纤维素基碳材料中,经先预碳化再活化方法,在800℃的活化温度下制备得到的P-800比表面积高达1737 m2/g,具有多孔结构以及丰富的缺陷;在6 mol/L KOH水溶液电解质中和电流密度0.5 A/g时,比容量达到194 F/g,并且具有良好的导电性能。
