UPLC-MS/MS法同时测定一清颗粒中5种成分的含量
2021-12-17黄丽杰崔燕贞李晓静
岳 磊 黄丽杰 崔燕贞 李晓静
1. 郑州市食品药品检验所,河南 郑州 450000;2.郑州大学第三附属医院,河南 郑州 450000
一清颗粒作为临床常用的中成药,在《中国药典( 2015 版) 》中规定,其处方为黄连、大黄和黄芩这三味药材,其功能与主治为清热、泻火、解毒、化瘀、凉血、止血[1]。现行的药典规定中,在含量测定项下使用高效液相色谱(HPLC)法,仅以黄芩苷作为目标物进行测定,不能有效的针对黄连及大黄进行有效的质量控制。近期关于一清颗粒质量控制的研究集中在制备工艺[2]、指纹图谱[3]及使用HPLC法检测更多的目标化合物[4],及针对其中大黄蒽醌类化合物的测定进而对其进行有效的质量控制[5-7]。本实验以超高效液相联合三重四级杆质谱法(UPLC-MS/MS)法来针对一清颗粒中的5种标识成分进行含量测定,质谱检测法可以在测定多种成分时,对被测成分在液相条件下的分离度没有强制要求,能具有更高的专属性和灵敏度。通过对一清颗粒中黄芩苷、盐酸小檗碱、大黄素、大黄酚及大黄素甲醚的液相条件和质谱条件进行优化,形成较为成熟的UPLC-MS/MS含量测定方法,可以在日常工作中帮助检验检测人员有效地提高检验工作时的工作效率和准确程度,为更全面地针对不同厂家,不同批次的一清颗粒的质量评价和控制提供技术支持。
1 仪器与试药
1.1 仪器 超高效液相色谱仪,型号:ACQUITY UPLC (美国Waters); 线型离子阱质谱分析仪,型号:4000 QTrap(美国Sciex);电子分析天平,型号:XS205DU(瑞士Mettler Toledo);超声波清洗机,型号:SB-800DTD(宁波新芝生物科技股份有限公司)。
1.2 试药 对照品均为中国食品药品检定研究院生产,分别为:黄芩苷(110715-201619,93.5%)、盐酸小檗碱(110713-201814,86.7%)、大黄素(110756-201512,98.7%)、大黄酚(110796- 201922,99.4% )、大黄素甲醚(110758-201817,99.2%);一清颗粒均为河南省及郑州市2018~2020年度安全监督抽检样品;乙腈、甲醇(德国Merck);甲酸(美国Fisher);纯净水(杭州娃哈哈公司)。
2 试验方法与结果
2.1 色谱与质谱条件
2.1.1 色谱条件 色谱柱:ACQUITY UPLC BEN C18(2.1 mm×50 mm,1.7 μm);流动相:乙腈(A)-水(含0.1%甲酸)(B),梯度洗脱程序(0~3 min,20%→90%A;3~4 min,90%A;4~4.5min,90%→20%A);流速:0.3 mL/min;柱温:30℃;进样量:1 μL。
2.1.2 质谱条件 电喷雾离子源(ESI);正、负离子实时切换模式;多重反应监测模式(MRM);源喷射电压:5500 V/-4500 V(随离子模式实时切换);源内温度(TEM):500 ℃;气帘气压力(CAD):20 psi;涡旋气压力(GAS1、GAS2):50 psi; 碰撞室气体:氮气。黄芩苷等5个成分具体质谱参数见表1。

表1 黄芩苷、盐酸小檗碱、盐酸黄柏碱、腺苷和大黄素的质谱参数
2.2 混合对照品溶液制备 精密称取5种对照品(黄芩苷、盐酸小檗碱、大黄素、大黄酚及大黄素甲醚)适量,分别置于适合的容量瓶中,分别取适量体积置100 mL量瓶中,加甲醇制成混合对照品溶液,对应各浓度分别为197.5362、52.0497、0.5116、0.5053、0.5071 μg/mL。
2.3 供试品溶液制备 取本品10袋,倒出混合后精密称定,研细,精密称取约0.1 g,置100 mL锥形瓶中,加入70%甲醇50 mL,精密称定。置超声机中满功率超声30 min,取出放至室温,称重后如有重量损失,使用70%甲醇补足其减失的重量,摇匀后使用0.22 μm微孔滤膜滤过,即得。
2.4 阴性对照溶液制备 取空白辅料,按药典一清颗粒制法项下的比例混合,制得阴性对照样品,按“2.3”项下的方法制得阴性对照溶液。
2.5 专属性试验 分别精密吸取稀释后的混合对照品溶液(精密量取混合对照品溶液5 mL,置50 mL量瓶中,加适量甲醇稀释,并定容至刻度,即得)、供试品溶液及阴性对照溶液各1 μL,按“2.1”项下的色谱条件与质谱条件进行进样测定,结果如图1所示。结果显示:供试品及对照品溶液在各成分的提取离子流图(XIC)中出峰时间一致,而阴性对照溶液在对应的XIC图中均没有干扰,其专属性强。
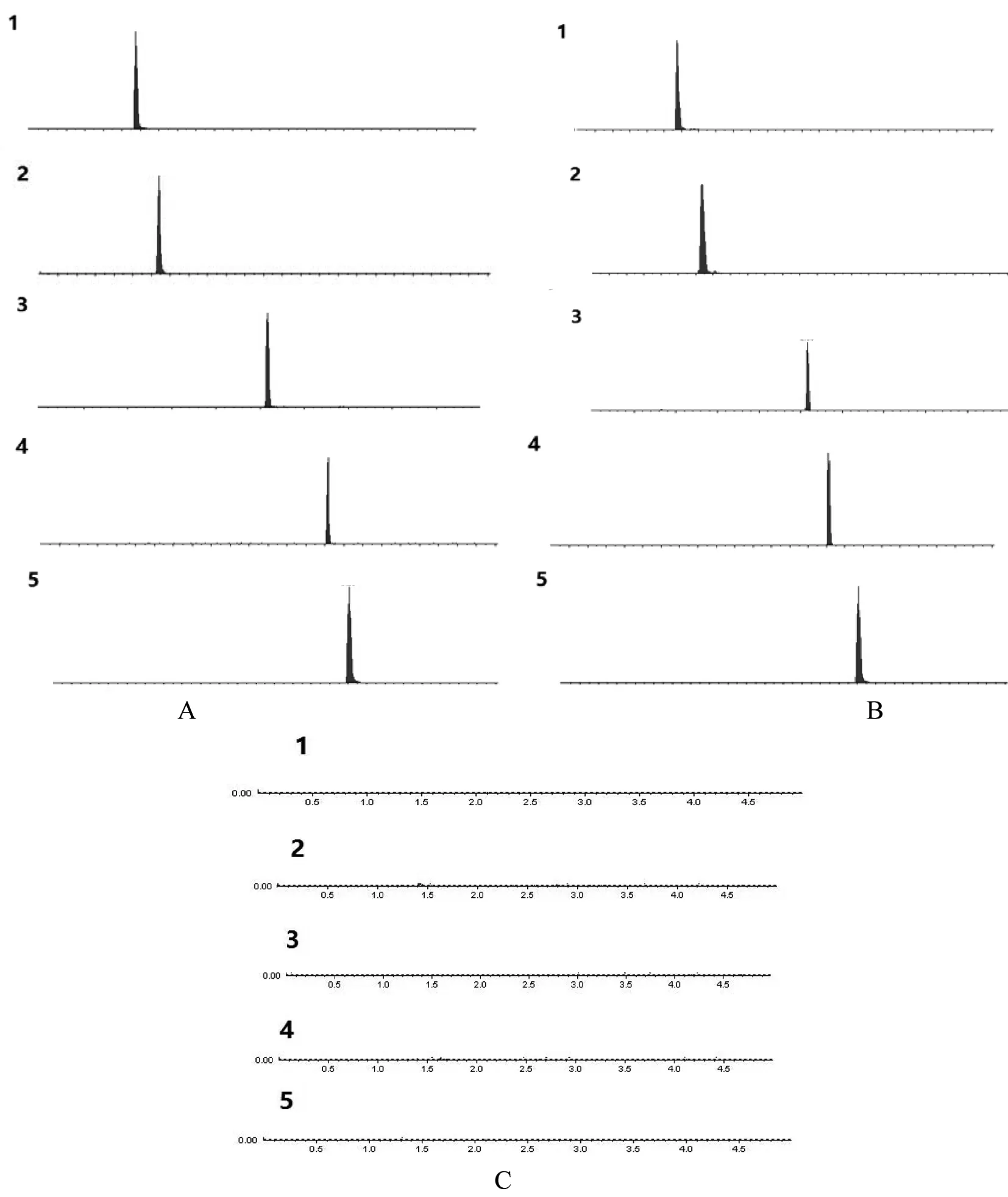
1.黄芩苷;2.盐酸小檗碱;3.大黄素;4.大黄酚;5.大黄素甲醚;A.对照品溶液; B.供试品溶液; C .阴性对照溶液图1 5种成分的提取离子流图
2.6 线性关系考察 精密量取“2.2”项下的混合对照品溶液1.00、5.00、10.00、50.00 mL,分别置100 mL量瓶中,用甲醇稀释并定容至刻度,摇匀即得。以各化合物的质量浓度为横坐标(X),以峰面积为纵坐标(Y)进行线性回归,分别以信噪比S/N=3、S/N=10时的浓度作为检测限和定量限,回归方程及相关系数见表2,各成分在对应范围内的线性关系良好。
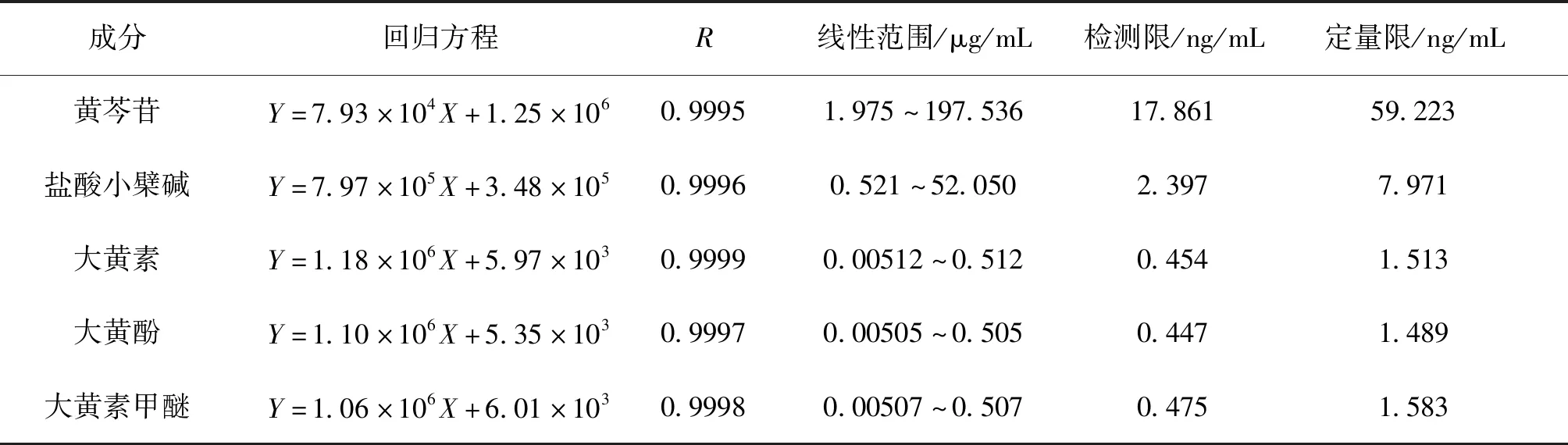
表2 线性关系、检测限和定量限
2.7 精密度试验 取“2.5”项下的稀释后的混合对照品溶液,取连续进样6次的数据分析。结果显示,黄芩苷、盐酸小檗碱、大黄素及大黄素甲醚峰面积的RSD分别为1.37%、2.21%、4.33%、4.49%、4.25%,显示本方法的精密度符合要求。
2.8 重复性试验 取编号为01的一清颗粒细粉各6分,平行制备6份供试品溶液,进样测定。测得各成分的平均含量分别为1.377、0.373、0.762、16.361、4.357 mg/片,其RSD分别为3.89%、4.18%、4.76%、2.32%、2.18%,显示本方法重复性符合要求。
2.9 稳定性试验 取同一份编号为01的供试品溶液,在室温下放置0、2、4、6、8、10、12h后测定。结果各成分的RSD分别为2.12%、2.38%、4.17%、4.42%、3.97%,表明该供试品溶液在12 h内具有较好的稳定性。
2.10 加样回收率试验 随机取同一批一清颗粒(编号为01),精密称取其颗粒细粉0.05g,精密加入混合对照品溶液适量,制备6份加标供试品溶液的平行样,计算加样回收率,结果显示,各成分的平均加样回收率分别为96.37%、95.15%、89.57%、90.64%、87.13%,其RSD分别为2.74%、2.19%、2.44%、2.56%、3.03% 。
2.11 供试品含量测定 全部样品共9批一清颗粒,按照“2.3”项下方法,分别制备供试品溶液进行分析,通过回归方程计算其5种成分的含量,结果见表3。
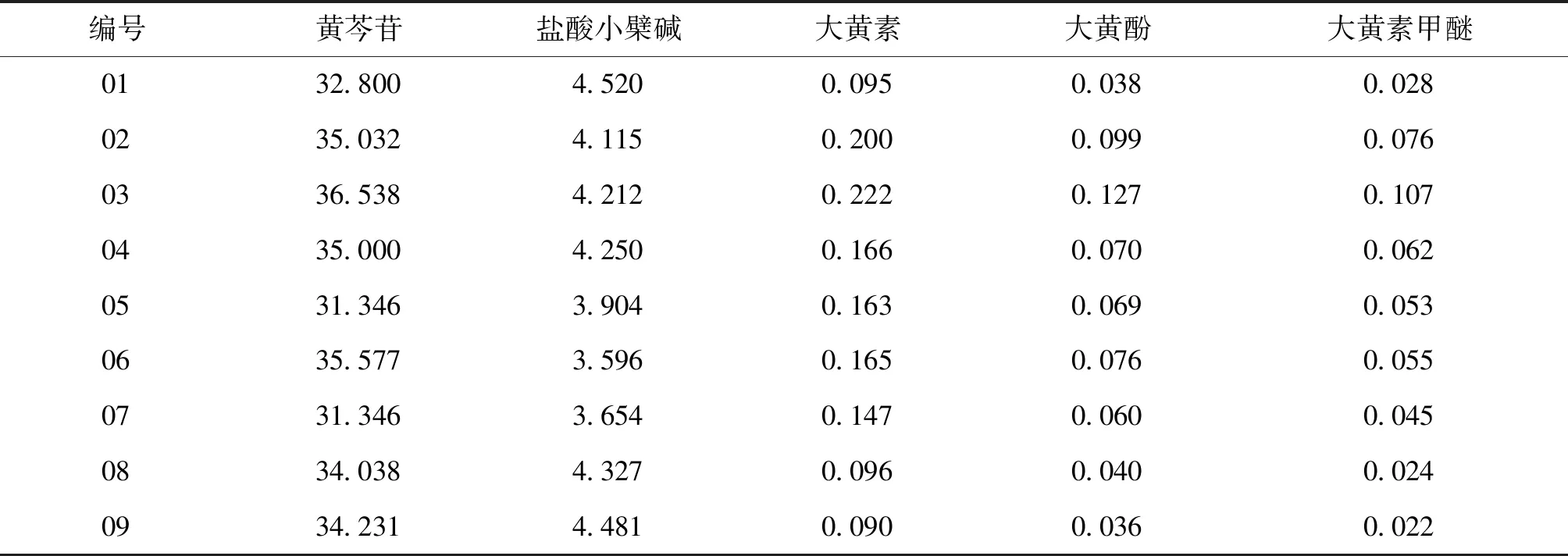
表3 供试品含量测定结果 (mg/包,n=2)
3 讨论
3.1 目标化合物的选择 通过药典记载可知,一清颗粒的处方为黄连、黄芩与大黄三味药材,通过分别煎煮后浓缩成浸膏粉,混合加辅料制成颗粒[1]。其中大黄的用量最大,不使用含量测定的方法对其进行质量控制显然不够严谨。沿用药典选择的黄芩苷作为黄芩的标志物的思路,实验中增加了盐酸小檗碱作为黄连的标志物,选用大黄素、大黄酚以及大黄素甲醚作为大黄的标志物进行含量测定,使用液质联用方法,一次性针对其中的目标成分进行检测,进而能够保证对其进行更为全面的质量控制。
3.2 提取方法的优化 针对供试品前处理中的提取方式、选用溶剂与提取时间进行了优化研究。其中提取方式选择了直接超声和回流提取,结果显示二者提取率差异较小,而超声提取操作较为便捷。选择的溶剂包括甲醇、50%甲醇、70%甲醇与90%甲醇,最终选用提取率较高的70%甲醇。超声提取时间在15 min、30 min与45 min之间进行对比,结果表明选用超声30min的样品,其提取率明显高于15 min,与45 min的样品相比差别不明显。经综合考量,最终确定选用提取溶剂为70%甲醇,超声提取时间选择30 min。
3.3 色谱条件的优化 流动相的优化先采用相同比例的甲醇-水、甲醇-水(含0.1%甲酸)、乙腈-水、乙腈-水(含0.1%甲酸)进行考察。结果显示使用乙腈作为有机相时,其图谱的峰型比甲醇的更好,目标化合物的分离程度也更好;而对比加入0.1%的甲酸的水相与纯水相,可明显看到加入0.1%的甲酸可以有效增强这5种化合物的信号强度,也能改善峰形的对称性。最后考察了不同梯度的洗脱方式,进一步提升了分离效率。最终选择乙腈-水(含0.1%甲酸)作为流动相,采用梯度洗脱方式进行试验。
3.4 结果分析 综上所述,本试验建立了使用UPLC-MS/MS法同时测定一清颗粒中5个成分的含量,9批样品测定结果中可以看出,黄芩苷作为药典方法的控制因素,其含量测定结果显示,各批次间差异较小,且均符合药典规定(每袋不得少于21mg)。盐酸小檗碱的含量测定结果,各个批次差异也相对较小;而以大黄素、大黄酚、大黄素甲醚作为大黄药材的特征成分来看,各批次间呈现出较大差异。
