电子探针定量分析影响因素浅析*
2021-11-15于凤云刘晓英李春艳
于凤云,刘晓英,李春艳
(大连理工大学材料科学与工程学院,辽宁 大连 116024)
电子探针(Electron Probe Microscope Analyser,EPMA)是通过聚焦电子束轰击样品表面,然后利用波谱仪测量试样受激发产生的特征X 射线波长及强度来确定被测样品微区化学成分的测试技术。电子探针具有分析区域小、准确度及灵敏度高、分析简便等特点,在材料科学、冶金、生物、地质、考古及轻工等领域应用广泛[1-2]。采用电子探针可对样品进行全元素、线扫描、面扫描等定性分析和定量点分析[3-5],其中,定量点分析是电子探针的核心功能,也是微区成分分析最重要的技术手段之一。本文针对业内关心的定量点分析测试准确度这一重要问题,分析了电子探针定量点分析的影响因素,并提出了实验过程中定量点分析出现较大偏差时的应对措施。
1 待测样品影响因素
1.1 待测样品表面平整度
待测样品表面存在的凹凸起伏会影响波谱仪对受激发特征X 射线的接收,造成测试结果产生偏差。样品表面凸起的影响主要体现为对特征X 射线的吸收,极端情况,当电子束进入一个凹陷的孔洞内时,特征X 射线甚至可以被完全吸收而无法检测[6]。表面凸起对特征产生额外吸收,如图1 所示。以日本电子电子探针(JXA-8530F PLUS)为例,取出角为40°。当测试位置附近存在较高凸起时,凸起部位会增加特征X 射线在物质中的穿行距离,产生二次激发,波谱仪接收到的特征X 射线波长和强度都会产生较大偏差[7]。
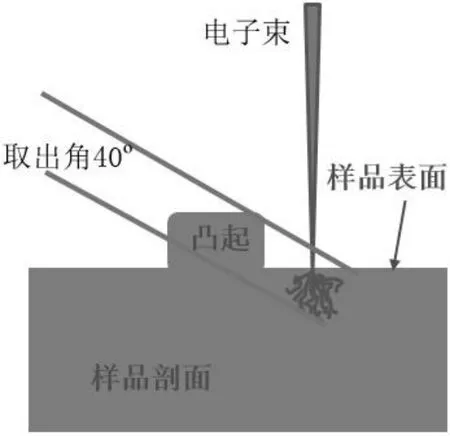
图1 表面凸起产生的额外吸收
样品制备应保证表面平整,具体操作可参考金相制样标准。如果样品本身存在较硬或较软的析出相,可在机械磨抛之后采用氩离子抛光仪精抛以提高样品表面质量。同时,实验中可以利用二次电子图像观察样品表面状况,视野中不应存在明显划痕,也可以通过监测吸收电流是否存在剧烈变化来评估样品的表面质量[8]。
1.2 样品导电性
当样品本身导电性较差时,电子束作用区域容易产生电荷聚集并形成干扰电场,造成电子束入射偏移和吸收电流的不规律波动,影响测试分析正常进行。对此类样品,测试前通常做导电处理,即在样品表面真空喷镀碳膜或其他导电金属薄膜。喷镀时要合理选择导电薄膜元素,一般要求选择与待测样品元素种类不同且本身密度小的元素。常规镀膜仪中主要包含碳和金两种元素,其中碳膜主要用于成分分析,而金膜导电性能更好,主要用于形貌观察。成分分析导电处理镀膜时,首先确保镀膜元素必须避开待测目标元素,以消除镀膜处理对分析结果的影响;其次是镀膜厚度不易过厚过薄,20~50 nm 较好。对样品做喷镀导电处理之后,入射电子束和激发特征X 射线在穿透镀膜时必定会损失部分能量,能量损失与膜厚度和测试电压有关,如图2 所示[8]。加速电压一定时,特征X 射线强度随镀膜厚度增加而降低;镀膜厚度一定时,加速电压越高,特征X 射线强度损失越小。目前国产标准样品表面均有一层碳膜,因此建议定量分析时在待测样品表面喷镀同等厚度的碳膜,改善样品导电性的同时也减小标准样品表面碳膜对定量分析结果的影响。
1.3 待测样品微区均匀性
待测样品微区均匀性也会影响电子探针定量分析结果的准确性。电子探针定量分析过程中先通过标准样品分析得到K因子,然后采用ZAF 校正法得到校正结果。当电子束直径范围内成分不均匀时,定量结果ZAF 校正是唯一值,不能真实反映样品真实基体情况,从而产生误差。如图3 所示,以Al-Si 样品为例,在Si 富集区,Si 与Si 之间的校正因子吸收因子A=1,无吸收。而校正过程按照元素均匀分布样品校正,而Si 元素的Kα特征X 射线默认被Al 吸收,故实际校正因子中吸收因子A>1,校正后测得的Si 元素浓度值偏高。因此,定量分析时样品分析区域成分必须均匀。
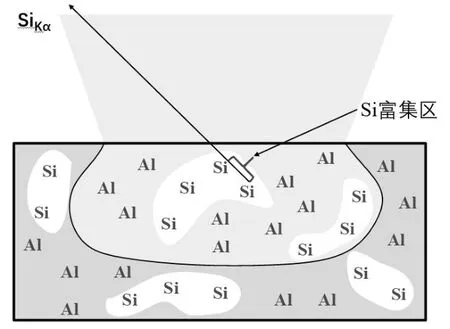
图3 元素不均匀样品示意图
1.4 待测样品元素迁移(样品损伤)
在大多数情况下,电子探针分析过程对样品不会造成损伤,因此电子探针分析通常被称为无损分析。然而对于碳酸盐、磷酸盐、含水富二氧化硅的玻璃[9]及含水碱金属铝硅酸盐玻璃[10]等无机盐样品,电子探针分析时电子束照射过程可能造成样品元素迁移,即样品损伤。样品损伤主要包含电子束激发区域中元素迁移造成的成分损伤和电子束辐照过程造成的样品局部热损伤。对于导热性差的样品,可在样品表面喷镀一层导热性好的薄膜,如Ag[11],或者降低束流,减少因测试区域热损伤而造成的定量分析偏差;在分析含K、Na、Al、Mg、Sr、Cl、O、N 等元素的样品时,可适当减小电流,尽量避免此类样品损伤导致的成分偏差[1]。
2 标准样品的影响
2.1 标准样品的要求
标准样品需具备成分已知、物理和化学性质稳定、在微米量级范围内成分均匀等特点。通常标准样品为某种元素的单质(纯金属),而对于化学性质较活泼的元素(碱族和碱土族元素)和常温下为气体的元素(N、O、F、Cl 等),其标准样品多为某种化合物。
2.2 标准样品键合状态
一些元素在化学结合或合金化时,被电子束激发出的特征X 射线波长发生改变而使谱线移位。这些谱线移位是由于电子的电屏蔽作用的改变而使能级发生变化所致。低原子序数的光谱中谱线移动量最大的谱线是能量最高的那条谱线[1]。如FeS、S[8]及Si[12]、Al、Mg 等氧化物与纯金属K 系谱线[13]均有不同程度移位。实验中存在谱线移位的元素应选择与待测样品元素化合价或配位数相同的标准样品。
2.3 标准样品氧化
纯金属标准样品(如Mg、Al、Ti、Mn、V 等)长期使用之后表面会形成一层致密的氧化膜,造成该元素的参考特征X 射线计数强度降低,导致定量分析结果偏高[14]。纯金属标准样品应定期打磨并真空存放,分析时为防止标准样品氧化,样品室中要保持较高的真空度。
3 测试过程的影响因素
3.1 电压束流
加速电压选择的主要考虑是X 射线激发效率和入射电子在试样中的穿透,激发效率与过压比U=Eo/Ec有关,Eo为入射电子能量,Ec为有关壳层的临界激发能量。由于特征X射线强度近似地按(U-1)1.67变化,为了得到更高的计数率,希望用更高的过压比U[7]。峰背比也随U而增加。但是随着Eo增加,电子穿透迅速增加,电子亦从其轰击点进一步散布,因而空间分辨率变差。此外,当电子穿透到较大的深度时,出射X 射线在试样中要穿行更长的路程,吸收校正就增加,由于校正值大时校正准确度减小,所以对于定量分析中不推荐使用太大的加速电压。提高电子束流也是获得高X 射线强度的有效方法,但电子光学系统的几何结构和参数限制束流不能太大,因为电子束流的增加将使电子束直径增加,从而降低了微区分析的空间分辨率[15]。
对于痕量元素分析时,应该选用较高的加速电压和电子束流。对于分析导电膜厚度不确定和容易碳污染的样品时应选择不低于10 kV 加速电压,因为使用过低的加速电压会导致X 线强度对导电膜和碳污染过于敏感。对于分析轻元素(Z<10)时,应选择较低的加速电压。
3.2 样品位置
波谱仪的分析原理为电子束入射到样品表面时,激发出携带样品成分信息的特征X 射线,经晶体分光聚焦后,被X射线计数管接收。电子探针分析过程样品测试位置必须保持在罗兰圆上[3]。偏离罗兰圆时波谱仪接收到的X 射线强度减弱,元素定量分析结果偏低。故电子探针成分分析时必须保证待测点光学聚焦清晰,此时待测点恰好位于罗兰圆上。
3.3 元素缺失
应用电子探针定量分析功能时,有测试者只关心对样品中某一种或某几种元素的准确含量,在检测过程中仅测量了样品中部分元素的含量。此时,由于测试过程中未准确获取ZAF 校正法所必须的基体信息,导致测试结果误差增大[16]。因此,在应用电子探针定量分析时应包含样品全部元素种类。
3.4 元素重叠峰
相比能谱仪EDS,波谱仪WDS 已经最大程度地降低了元素间的重叠峰干扰,但实际测试中仍会因元素重叠峰干扰而造成定量分析结果偏差,特别是对于样品中微量轻元素,其测量偏差更大[17]。波谱仪WDS 分析中的干扰峰主要体现为高阶峰对低阶目标峰的影响。以含Ti、N 样品为例,Ti元素的TiLL高阶峰会对N 元素的K 阶主峰NKα形成干扰,如图4 所示[18]。待测N 元素在K 阶主峰NKα处的实测强度值由两部分组成,分别为Ti 元素在峰位的干扰峰强度和 N 元素在 K 阶特征峰 NKα的净强度[18],即:
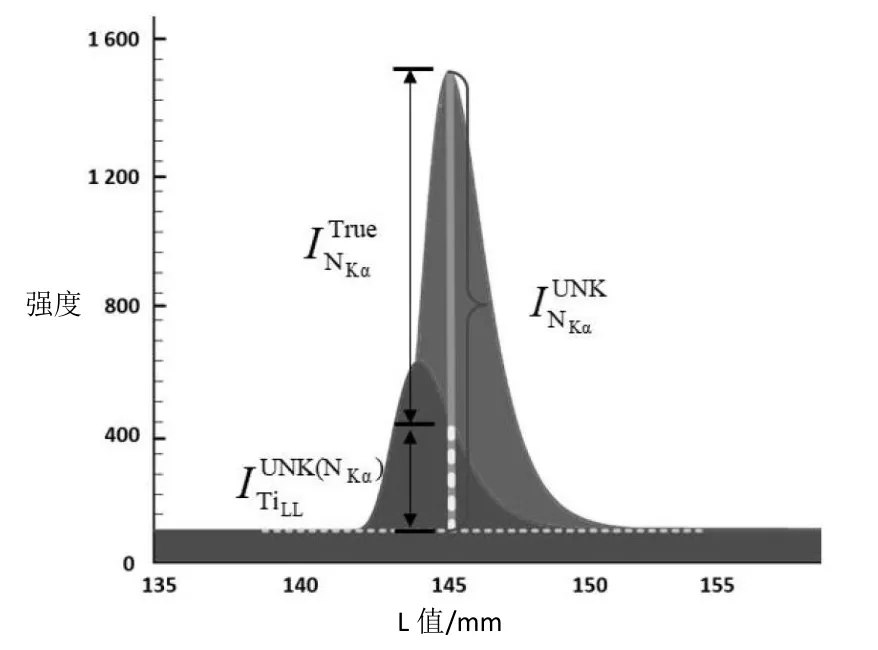
图4 Ti 元素高阶峰TiLL 对N 元素主峰NKα检测强度的干扰图[18]
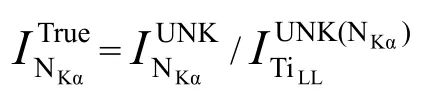
Ti、N 元素之间的重叠峰干扰对N 元素含量的定量分析会产生较大影响,须从待测样品的实测强度中去除重叠干扰峰相应的强度[19],从而获得N 元素的真实值。
3.5 电子束直径及电子束扫描方式
电子束直径是通过电子束散焦实现。电子束直径变大时,样品只有位于电子束中心点位置处满足在罗兰圆上的要求,而样品其余区域位置则偏离罗兰圆,如图5 所示。电子束直径越大,偏离罗兰圆的面积在电子束照射范围内的比例越高,定量分析结果偏差也越大。
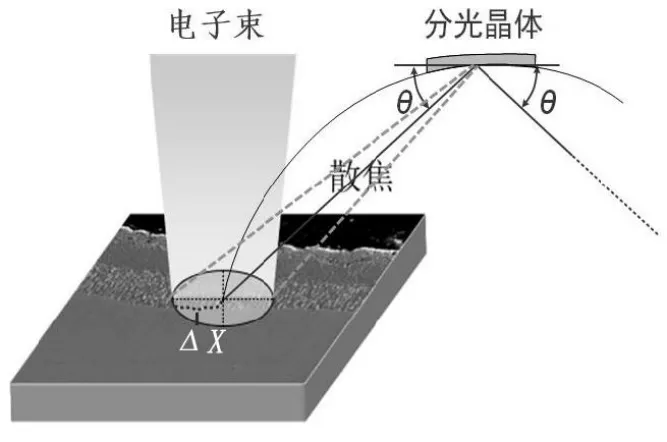
图5 电子束散焦时与罗兰圆的位置关系
定量点分析时,测试标准样品电子束直径与待测样品电子束直径差最好不超过20 μm。定量分析时分析区域边长小于20 μm 时可选用电子束扫描模式测试元素含量。
3.6 环境温度
电子探针分光晶体的晶面间距会随环境温度的变化发生改变,导致谱线位置发生偏移。对部分峰宽较小的元素,谱线峰位发生微小改变时,其测量结果就会产生显著偏差。特别是对于LiF 晶体,其环境温度变化超过±3 ℃时,就会产生明显的峰值移位和强度改变。待测样品和标准样品测量时环境温度的差异会造成较大误差。在电子探针分析中,环境温度变化引起谱线移动偏移,可等于或超过由于化合价或配位数变化而产生的谱线移动[1]。这一误差的解决办法是保证实验室温湿度恒定,同时在试验前对标准样品进行标定,避免使用系统中的标准样品数据。
3.7 碳污染
样品表面往往会吸附环境中的碳氢化物,电子束激发待测样品时碳氢化物发生裂解,产生游离碳并沉积于样品表面。电子束和特征X 射线在穿透碳沉积层时会损失部分能量,降低特征X 射线的强度,从而导致定量分析结果偏低[7]。预防的办法可用一冷指,或者用一气嘴。冷指的作用是使蒸汽在与电子束交互作用前就沉积。而低压空气喷嘴与电子束的离化能力相配合可不断地除去因轰击溅射产生的污染膜。解决这一问题的方法是设法排除真空系统中的碳氢化物,实验过程中可以通过提高样品表面清洁度,减少碳污染。
3.8 背底校正
连续X 射线谱是背底的主要来源。准确的背底扣除对于电子探针分析是至关重要的,对微量元素的定量分析尤为明显。选择背底位置扣除背底时,首先背底位置选择要避免可能存在的一阶或更高阶的干扰峰位置。其次在确定待测特征峰背底从一侧过渡到另一侧时,背底连续谱线必须与两侧背底的形状/斜率保持一致。大多数情况波谱仪测量背底比较简单,只需把波谱仪转到谱峰两侧进行测量,用线性内插算出峰下面的背景值[20],如图6(a)所示。但并非所有背底都是线性的,当波谱仪晶体距样品最短的距离处需要注意,背景显示出增强的向上弯曲,从而产生异常高的背景[21],如图6(b)所示。在这种情况下,必须使用弯曲的背底模型来准确确定背底[16]。
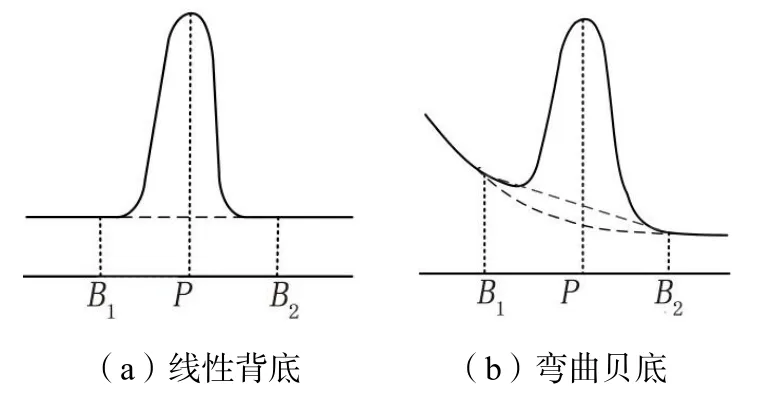
图6 根据背底B1、B2 位置的强度计算峰位置P 处的背底强度
3.9 氧化物定量分析
目前氧化物定量分析方法常用有两种:金属ZAF 法和氧化物ZAF 法。氧化物法是通过测试样品中阳离子,配算出氧元素含量的一种测试方法,该方法可以提高定量分析结果的精度,减小误差。若待测样品存在氧化物且同一元素是单一价态时,推荐选用氧化物ZAF 法;若待测样品氧化物同一元素无固定价态时推荐采用金属ZAF 法进行定量分析。
4 结语
电子探针定量点分析能够快速获得块体样品中的微区成分信息,但定量分析结果的影响因素较多,测试操作不当时往往会造成较大的测量偏差,甚至给出错误结果。电子探针定量分析中较为理想的元素浓度总量应在99%~101%之间。定量分析结果元素浓度总和不在98%~102%范围内时,需逐一筛查可能的影响因素,消除干扰,提高结果准确度。
