界面化学对镍锰酸锂正极材料电化学性能影响的研究现状及分析
2021-11-14廖文俊
周 兰,李 旺,廖文俊
(上海电气集团股份有限公司中央研究院,上海200070)
近年来,对能源的巨大需求激发了高能量密度锂离子电池材料的发展,尤其是电动汽车领域,目前商业化的锂离子电池仍无法满足电动汽车对电池低成本及高能量密度的要求。尖晶石型的LiNi0.5Mn1.5O4正极材料因其价格低廉、对环境无污染、安全性能好、高能量密度等优点而成为当今锂离子电池正极材料研究的热点之一。其理论放电比容量可达146.7 mA·h/g,电压平台高达4.8V,具有超高的能量密度(650 W·h/kg)及功率密度,目前已经引起广泛关注,被认为是未来锂离子电池发展中最有前景的锂离子电池正极材料之一[1-3]。本文从LiNi0.5Mn1.5O4正极材料的结构及脱嵌机理入手,对近年来国内外在LiNi0.5Mn1.5O4正极材料和电解液方面的研究进展进行总结和分析,着重探讨了界面化学对LiNi0.5Mn1.5O4正极材料电化学性能的影响情况,特别是近几年围绕LiNi0.5Mn1.5O4正极材料表面和电解液界面提出的新方法和新思路,并对今后的研究和发展趋势进行了展望。
1 LiNi0.5Mn1.5O4正极材料的结构及脱嵌机制
LiNi0.5Mn1.5O4存在非化学计量比无序和化学计量比有序两种尖晶石结构,分别对应Fd3m和P4332两种空间群,如图1所示[4],在Fd3m中,Ni和Mn原子在进行占位时,是随机分布在16d位置,Li原子位于8a位置,O原子位于32e位置;而在P4332中,Ni原子和Mn原子有序地排列在镍锰酸锂晶格结构中,Ni和Mn原子分别占据4a和12d位置,Li原子占据8c位置,O原子分别占据24e和8c位置。
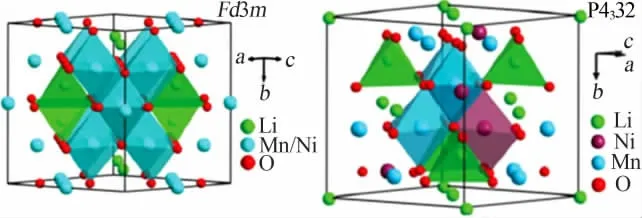
图1 尖晶石型正极材料LiNi0.5Mn1.5O4结构示意图[4]Fig.1 Structural diagram of spinel LiNi0.5Mn1.5O4 cathodes[4]
P4332有序型结构LiNi0.5Mn1.5O4中的Mn为+4价,在实际制备过程中,可以通过改变煅烧工艺或掺杂改性等方式向材料中引入微量Mn3+,为了保证材料的电中性,材料结构中会出现氧空缺,使得材料的空间群由P4332逐渐变化为Fd3m,这种结构的改变也被称为LiNi0.5Mn1.5O4材料的无序化过程,无序程度不同Mn3+的含量也不同。通常认为,Fd3m型的LiNi0.5Mn1.5O4材料相比于P4332型具有更优异的倍率性能,这与晶胞参数大小及锂离子的扩散系数有关,无序型Fd3m结构更有利于Li+和电子的传输,因而具有更好的导电性。有序型和无序型LiNi0.5Mn1.5O4可以通过XRD、红外等多种方式判定,其中充放电曲线是最典型的判定方式之一,如图2所示[5]。

图2 有序型与无序型尖晶石LiNi0.5Mn1.5O4正极材料充放电曲线对比图[5]Fig.2 Comparison diagram of charge and discharge curves of ordered and disordered LiNi0.5Mn1.5O4 spinel cathodes[5]
对于P4332型有序LiNi0.5Mn1.5O4材料,Mn完全为+4价,Ni为+2价,充放电过程中,只在4.7 V左右出现了单一电压平台,对应Ni2+/Ni4+的氧化还原反应过程;而对于Fd3m型无序LiNi0.5Mn1.5O4材料,在4.6 V左右和4.8 V左右出现了两个平台,分别对应Ni2+/Ni3+和Ni3+/Ni4+两个过程,且由于微量Mn3+的存在,在4.0 V附近出现了Mn3+/Mn4+的小平台[6]。
2 表面和界面化学
对于镍锰酸锂电池来说,各种副反应,包括高电位下电解液的分解、电极的腐蚀、金属Mn、Ni的溶解及高活性Ni4+对电解液反应的催化等问题会导致活性材料的流失及电池阻抗的增大,最终使得电池库伦效率降低、循环性能变差。而对于锂离子电池来说,电子迁移和锂离子扩散通常发生在活性材料表面以及与其他物质组成的各种界面之间,因此通过改善LiNi0.5Mn1.5O4材料的表面和界面化学对于提高镍锰酸锂电池的电化学性能有着至关重要的作用。
2.1 LiNi0.5Mn1.5O4表面化学
2.1.1 晶面取向
对于LiNi0.5Mn1.5O4材料来说,不同的晶面取向,不仅决定了其晶体形态的不同,更对其电化学性能有着重要的影响[7-13],这主要与Li+在LiNi0.5Mn1.5O4中扩散的各向异性动力学有关。例如HAI等[7]曾报道过板面形状和八面体形状的LiNi0.5Mn1.5O4,并对其不同的化学扩散系数进行了深入研究,结果表明八面体结构的LiNi0.5Mn1.5O4相比于板面结构的LiNi0.5Mn1.5O4则具有更高的容量和更好的倍率性能,这主要是八面体结构的LiNi0.5Mn1.5O4中对应的(111)晶面相比于板面结构的LiNi0.5Mn1.5O4中对应的(112)晶面具有更高的Li离子扩散系数。DENG等[8]利用固相法分别制备了P掺杂的八面体和截角八面体的LiNi0.5Mn1.5O4材料,如图3所示。通过不同比例P掺杂向材料中引入不同量的Mn3+而使得LiNi0.5Mn1.5O4无序度增加的同时也对晶面形态产生了影响,如LiNi0.5-xP2xMn1.5-xO4中x分别为0和0.005时,晶体形态为正常的八面体形状,仅具有(111)晶面,而当x逐渐增大为0.01和0.02时,晶体形态变为截角的八面体形状,并伴随着(111)晶面的减少及(100)晶面的增多。电化学结果表明,LiNi0.5Mn1.5O4材料掺杂P后材料的无序度增加,由于无序型Fd3m结构更有利于Li+和电子的传输,而使得材料的电化学性能大大提升;但随着P掺杂量的进一步增加,晶体形态也发生了转变,(100)晶面不断增多而(111)晶面不断减少,这时晶面取向开始占据主导地位,这时(111)晶面的大幅度减少会对Li+扩散产生不利影响,因此随着P掺杂比例的进一步提高,材料的电化学性能下降。当然,CHEMELEWSKI等[9]也在文献中进一步证实(111)晶面与电解液之间具有更优异的界面稳定性。但是,近年来也有一些其他观点出现,CHEN等[13]通过液相可控法合成出了具有(111)晶面外还同时拥有(001)、(110)、(113)、(103)等晶面的类球形截角多面体结构,相较于仅有(111)面的八面体结构,这种截角多面体结构使得LiNi0.5Mn1.5O4材料具有更优异的容量保持率和倍率性能,这种性能的提升被归功于(111)面的减少和(110)面的增加,CHEN等[13]认为(110)面更有利于锂离子扩散。不仅如此,(111)面的减少还能有效抑制Mn的溶解,从而避免由于Mn3+的溶解而导致Jahn-Teller效应的发生,大大提高了材料的稳定性。LIU等[14]通过微波水热法合成出具有(100)面的截角八面体结构的镍锰酸锂材料,并表现出相当不错的倍率性能及长 循 环 性 能,因 此LIU等[14]认 为(100)面 相 较 于(111)面更有利于LiNi0.5Mn1.5O4材料电化学性能的提升。尽管如此,对于尖晶石体系来说,究竟是(111)面还是(100)面对电化学性能提升作用大仍存在争议。笔者认为之所以会出现这两种观点,是因为对于LiNi0.5Mn1.5O4材料来说,电化学性能的好坏不仅与晶面取向有关,同时还与颗粒形貌、阳离子Ni2+和Mn4+在晶格中的排序、岩盐相LixNi1-xO杂质的形成、Mn3+的含量、氧缺陷等众多因素有关,在设计材料的多面体结构过程中,这些因素中的一项或多项也在跟着发生改变,从而无法准确判断出电化学性能的好坏是否只是因为晶面结构的变化所导致。

图3 高倍透射电镜图(a1~d1,a2~d2)[8]Fig.3 HRTEM images(a1~d1,a2~d2)[8]
2.1.2 颗粒形貌
LiNi0.5Mn1.5O4合成方法主要有固相法、共沉淀法、溶胶凝胶法、喷雾热解法、水热/溶剂热法与熔盐法等。不同的合成路径、特定添加剂和表面活性剂将直接影响晶体生长,形成不同形貌。与微米级的LiNi0.5Mn1.5O4颗粒相比,纳米尺寸的LiNi0.5Mn1.5O4具有更优异的倍率性能,但从循环稳定性的角度来说,纳米颗粒虽然能够有效缩短Li+的扩散路径,提高材料的倍率性能,但其形成的大比表面会大大增加与电解液的接触面积,加剧与电解液的副反应并增加过渡金属离子的溶解,因此亚微米尺寸结合三维或多孔结构等特殊形貌的LiNi0.5Mn1.5O4材料目前研究的更多,效果也更好。LIU等[15]通过静电纺丝结合退火处理的方法制得直径为200~500 nm、长度为几十微米的纤维结构,这种三维结构可以实现充放电过程中Li+和电子的快速传输,具有良好的电化学性能,在0.5C时,其初始放电容量为133 mA·h/g,30次循环后容量保持率超过94%。HAO等[16]采用低温两步水热法成功得到亚微米尺寸的八面体形貌材料,表现出良好的倍率性能,1C时其放电比容量接近130 mA·h/g,5C时比容量仍接近100 mA·h/g。
2.1.3 表面元素分布
研究表明LiNi0.5Mn1.5O4表面的元素分布对其电化学性能也有很大影响,随着尖晶石LiNi0.5Mn1.5O4中Mn3+含量的增多,通过歧化反应产生的Mn2+溶解到电解液中的量也会增多,因此降低LiNi0.5Mn1.5O4表面Mn3+的含量可以有效减少Mn2+向电解液中的溶解,然而Mn3+对LiNi0.5Mn1.5O4电化学性能的影响一直存在争议。罗英等[17]研究发现通过优化Mn3+含量,控制材料形貌可以实现高性能LiNi0.5Mn1.5O4材料的制备。XIAO等[18]通过原子模拟指出,Mn3+的形成促进了Ni-Mn位点的无序排列,这种无序排列更有利于Li+的传输,尤其是大倍率充放电的情况下表现更为明显。而且,由于Mn3+含量的增加所带来的电子跳跃使得无序(Fd3m)尖晶石LiNi0.5Mn1.5O4的室温电导率比有序(P4332)高出2.5个数量级,这对高温下容量保持率是有益的。未来还需要进一步优化出Mn3+的最适比例,以此来平衡Mn3+对LiNi0.5Mn1.5O4电化学性能的双重影响。
除了上述Mn元素影响外,Ni元素对LiNi0.5Mn1.5O4材料的电化学性能影响也很大。LIU等[19]研究发现,55℃下循环后的LiNi0.5Mn1.5O4电极出现了材料的粉化现象,归咎于Ni促进了电解液的氧化分解,并在正极表面形成了钝化层(CEI),从而阻碍了Li+的扩散,最终影响了循环性能。而Cr掺杂后的LiNi0.5-xCr2xMn1.5-xO4(0≤2x≤0.8),由于Cr3+部分取代了Ni2+和Mn4+,使得材料表面跟电解液接触的Ni含量减少,这有利于电极/电解液的界面稳定性,降低了表面的阻抗,提高了镍锰酸锂材料的循环性能。LEE等[20]选用Cr-,Fe-和Ga-取代Ni-制备LiMn1.5Ni0.5-xMxO4(M=Cr,Fe,Ga)材料,也得出类似的结论。他们认为掺杂后的材料具有更稳定的电极/电解液界面,可以显著提高材料的循环和倍率性能。原因是这几种掺杂离子具有一定的自偏析效应,TOFSIMS深度测试结果如图4所示。由图4可以看出[20],与Ni相比,掺杂离子更容易富集在材料表面而造成表面贫Ni的状态,进而有效抑制了电解液的氧化分解。

图4 LiMn1.5Ni0.5-xMxO4(M=Cr,Fe,Ga;x=0、0.08)样品的飞行时间二次离子质谱(TOF-SIMS)深度剖析分布图[20]Fig.4 Time of flight secondary ion mass spectrometry(TOF-SIMS)depth profifiles of the LiMn1.5Ni0.5-xMxO4(M=Cr,Fe,Ga;x=0、0.08)samples[20]
2.1.4 表面结构的转变
充放电过程中表面结构的变化很难直观表征出来,因此关于表面结构的转变对LiNi0.5Mn1.5O4电化学性能的影响鲜有报道。LIN等[21]通过STEM发现,与没有充放电的原始LiNi0.5Mn1.5O4材料相比,充电到4.9V的LiNi0.5Mn1.5O4材料表面有约为2nm厚的类Mn3O4结构的薄层,与四面体的锂位被过渡金属离子部分占据有关。亚表层区域出现了岩盐结构,由八面体空位被过渡金属离子部分占据而引起,可以推断出的是充电后的LiNi0.5Mn1.5O4材料中过渡金属离子占据锂位会阻碍Li+的扩散,导致电荷转移阻抗增加,从而对LiNi0.5Mn1.5O4材料容量衰减以及首圈库伦效率产生一定的影响。研究针对LiNi0.5Mn1.5O4材料中表面化学与电化学性能的关系从原子层面出发给出了基本的认知并拓宽了研究领域,就如何稳定镍锰酸锂材料的结构和提高其电化学性能的问题给出了建议,即将尖晶石LiNi0.5Mn1.5O4表面的四面体Li位被少量不溶性离子预先占据。当然,为了进一步理解充放电过程中表面结构的转变对LiNi0.5Mn1.5O4电化学性能的影响,未来可以通过同时分析界面阻抗的变化、Mn离子的溶解、电极/电解液界面的稳定性来实现。
2.2 LNMO/电解液界面化学
2.2.1 电解液分解
镍锰酸锂材料充电到5 V高电位下,电解液易受材料中高氧化态过渡金属离子Ni4+的诱导而发生分解,并且溶剂和电解质都会参与反应,反应产物沉积在正极/电解液界面层形成表面膜(简称CEI膜)。传统碳酸酯类电解液由于LiPF6的存在,会发生分解形成PF5与LiF,由于微量水的存在,PF5又会转变为LixPOyFz,而溶剂EC与DMC会被氧化形成聚醚与聚碳酸酯。因此,CEI膜主要由聚碳酸酯、聚醚等有机组分及LiF、LixPOyFz等无机组分构成。研究发现,电解液的降解产物形成的这层CEI膜并不能均匀包覆在正极材料表面,因此不能有效地保护LiNi0.5Mn1.5O4正极。而且,随着循环的进行,电解液持续被反应,LiNi0.5Mn1.5O4正极上的表面膜不断增厚,往往会导致电池阻抗持续增加及容量的衰减。
为了进一步改善LiNi0.5Mn1.5O4的循环性能,越来越多的研究人员开始关注高电压电解液。一方面,开发耐5 V高电压电解液,希望大大突破商业化电解液4.3 V的瓶颈;另一方面,开发新型添加剂,在正极表面更好地参与成膜反应,防止电解液与正极材料的直接接触,进一步抑制电解液分解。在新型电解液体系方面,张文林等[22]合成了功能化离子液体1-丁基-3-甲基咪唑双(三氟甲磺酰)亚胺盐(BMIMTFSI)作为高压锂离子电池电解液组分,用于抑制有机溶剂的氧化,以提高碳酸酯类电解液的耐高压性。结果表明,当在电解液中添加20%(体积分数)BMIMTFSI时,LiNi0.5Mn1.5O4/Li电池在室温0.2C下的最高放电比容量是126.81 mA·h/g,5C下的放电比容量为109.36 mA·h/g,比在1 mol/L LiPF6-EC/DMC电解液中的放电比容量提高了91.7%,且该电池在0.2C下循环50圈后的放电比容量保持率在95%左右,比用碳酸酯类电解液提高了近10%。在新型添加剂方面,XU等[23]在碳酸酯基电解液中分别加入GBL(γ-丁内酯)和GLN(戊二腈)作为电解液添加剂,优先于溶剂分子发生氧化/还原分解反应,提高了电解液的氧化分解电压,并在电极表面形成一层有效的保护膜,可抑制碳酸酯基溶剂的后续分解,提高电极/电解液界面稳定性,整体优化电池性能。
2.2.2 过渡金属离子的溶解
金属离子溶解是导致材料容量衰减的另一原因。 当LiNi0.5Mn1.5O4在高压下工作时,由于电极 表 面 的 歧 化 反 应[24](2Mn3+→Mn2++Mn4+)以 及 电极和电解液之间的催化分解反应[25-27](由于微量水的存在,电解液中含有少量的HF,分解反应为:2Li Ni0.5Mn1.5O4+4H++4F-→3Ni0.25Mn0.75O2+0.75MnF2+0.25NiF2+2LiF+2H2O),导致过渡金属离子的溶解。PIECZONKA等[25]研 究 了Mn和Ni在 不 同 条 件(如荷电状态、温度、存储时间)下的溶解行为,发现LiNi0.5Mn1.5O4中Mn和Ni的溶解量随着SOC(剩余电量)、温度和储存时间的增加而增加。这是因为乙醚是碳酸二乙酯(DEC)在电解液中的分解产物,随着SOC、温度和贮存时间的增加而增加。在乙醚的形成过程中(通过DEC的另一种分解产物乙醇的脱水反应)会产生额外的氢氟酸,加速了LNMO中锰和镍的溶解。最近,JARRY等[27]提出了双质子耦合电子转移(PCET)反应机制用来描述LiNi0.5Mn1.5O4和碳酸酯电解液之间的界面反应过程,包括碳酸酯的电化学氧化及电极/有机电解液界面上锰和镍的溶出,通过X射线、荧光光谱及成像实验等表征手段证明碳酸酯在大于4.2 V的电压下会发生电化学氧化,导致Ni2+、Mn2+/3+与β-二酮配体结合及Ni2+、Mn2+与碳酸根配体结合形成各种金属络合物,从而引起Ni/Mn的溶解,且随着氧空位的增加,这种络合反应的速率也会加快。要想减少这种界面反应的发生,降低过渡金属的溶解,目前也有一些解决策略,比如可以优化正极材料的形貌、颗粒大小、晶面结构、包覆和掺杂等,电解液方面则可以通过优化溶剂、添加剂、离子液体等方式开发耐高压的电解液。
3 LiNi0.5Mn1.5O4正极材料的改性研究
大量研究表明,通过表面包覆和离子掺杂可以有效解决上述表面及界面化学带来的问题,极大提高电极材料的电化学性能。
3.1 表面包覆
考虑到电解液在高温下更容易分解,并且分解产物并不能均匀包覆在正极材料表面,因此不能有效地保护LiNi0.5Mn1.5O4正极。而且随着循环的进行,电解液持续被反应,LiNi0.5Mn1.5O4正极上的表面膜不断增厚,生成的副产物还会加速Mn的溶解,导致电池 阻 抗 持 续 增 加 及 容 量 的 衰 减。PANG等[28]对LiNi0.5Mn1.5O4分别进行了SiO2和聚酰亚胺包覆,如图5所示。由图5研究表明,包覆对有序的LiNi0.5Mn1.5O4性能提升比无序的更明显。除此之外,SiO2包覆后的LiNi0.5Mn1.5O4材料与未包覆的材料在25℃和55℃下的性能相比,容量衰减方面分别减少了45%和65%,较包覆之前容量衰减率明显降低,循环性能大大提升;聚酰亚胺包覆的LiNi0.5Mn1.5O4材料在常温下对性能的提升并不明显,但高温(55℃)下较包覆之前具有更低的容量衰减率,循环性能甚至优于SiO2包覆的LNMO材料,说明聚酰亚胺包覆层对材料的高温稳定性更有利,这与聚酰亚胺优异的机械性能和化学稳定性相关。总之,包覆可以有效隔离电极和电解液,显著改善材料的循环性能,选择不同的包覆物对性能的提升程度至关重要。除了上述提到的氧化物、高分子聚合物外,目前包覆比较多的还有磷酸盐[29-30],氟化物[31-32]等,虽然一定程度上改善了LiNi0.5Mn1.5O4材料的循环性能,但这类包覆层往往是电子与离子的不良导体,可能会对材料的倍率性能造成一定的负面影响,因此更多的研究者在努力寻找更为合适的包覆材料。

图5 原始LNMO的FE-SEM图(a1,a2)、SiO2-包覆LNMO的FE-SEM图(b1)和TEM图(b2)、PI-包覆LNMO的FE-SEM图(c1,c2)Fig.5 FE-SEM images(a1,a2)of original LNMO,FE-SEM image(b1)and TEM image(b2)of LNMO coated with SiO2、and FE-SEM images(c1,c2)of LNMO coated with PI
3.2 离子掺杂
离子掺杂可以有效改善LiNi0.5Mn1.5O4正极材料的性能,目前研究最多的是包括Al[33-34]、Mg[35]、Fe[36-37]、Zn[38]、Cu[39]、Co[40-41]、Cr[42]等金属离子在内的阳离子掺杂,通常金属阳离子取代部分Ni或部分Mn,有以下几点作用:1)抑制LixN1-xO等杂相生成;2)增大晶胞参数,有利于离子和电子传输;3)可诱导材料结构发生转变(由P4332型转变为性能更优的Fd3m型)。最近,LEE等[20]的研究表明,选用Cr-,Fe-和Ga-取代Ni-制备LiNi0.5-xMn1.5MxO4(M=Cr,Fe,Ga)材料,由于这几种掺杂离子具有一定的自偏析效应,与Ni相比,更容易富集在材料表面而造成表面贫Ni的状态,进而有效抑制电解液的氧化分解,从而改善材料的循环性能。除了阳离子掺杂外,目前也有一些阴离子掺杂来改善LiNi0.5Mn1.5O4性能的报道,多以F或S等阴离子取代部分O。以F元素掺杂为例,OH等[43]采用喷雾干燥法,以LiF为F源,制备的LiNi0.5Mn1.5O4-xFx(0 图6 Cr-Al-F掺杂前后的LiNi0.5Mn1.5O4材料分别在1C下的循环性能Fig.6 Cycle performance of LiNi0.5Mn1.5O4 materials before and after doping with Cr-Al-F at 1C 尖晶石型锂离子电池正极材料LiNi0.5Mn1.5O4具有高工作电压平台、高比容量、高能量密度、环境友好、循环性能好、三维锂离子扩散通道等优点,在未来电动汽车、化学电池储能等领域具有巨大潜在应用价值。但是,高工作电压带来的表面/界面等化学问题,大大制约了该材料的实际应用。本文从LiNi0.5Mn1.5O4正极材料的结构及脱嵌机理入手,对近年来LiNi0.5Mn1.5O4材料的研究进行了分析和总结,着重探讨了表面/界面化学对LiNi0.5Mn1.5O4正极材料电化学性能的影响情况,旨在对LiNi0.5Mn1.5O4正极材料表面改性和电解液界面构筑方面提出一些思路。 1)电解液分解是目前制约LiNi0.5Mn1.5O4应用的最主要瓶颈,寻找耐高压的电解液是关键,考虑到电解液与正极材料的兼容性,溶剂和添加剂的选择未来还需要不断优化。 2)正极材料的表面处理也起着重要的作用,离子掺杂与表面包覆可显著改善材料的电化学性能,未来需要寻找更合适的掺杂离子,更优的包覆物,或者将掺杂和包覆结合起来,取长补短,利用两者的协同效应,更有效地提高材料的电化学性能。 3)包覆方法上,也可以利用电解液添加剂在电极表面的优先分解对正极材料实现原位包覆,对包覆层的厚度及主要成分需要进一步研究。当然,也可以在正极材料表面构造浓度梯度,或者掺杂具有自偏析效应的金属离子,使正极材料表面处于贫镍状态,尽可能地减少与电解液的副反应,或者采用不同的合成方法,控制晶面取向向电化学性能有利的方向生长等。 未来希望在提高LiNi0.5Mn1.5O4正极材料电化学性能的同时,能够进一步探讨其工作机理,为人们深入研究和开发LiNi0.5Mn1.5O4正极材料提供一定的指导依据,这些将有利于加速高比能量锂离子电池正极材料的商业化进程。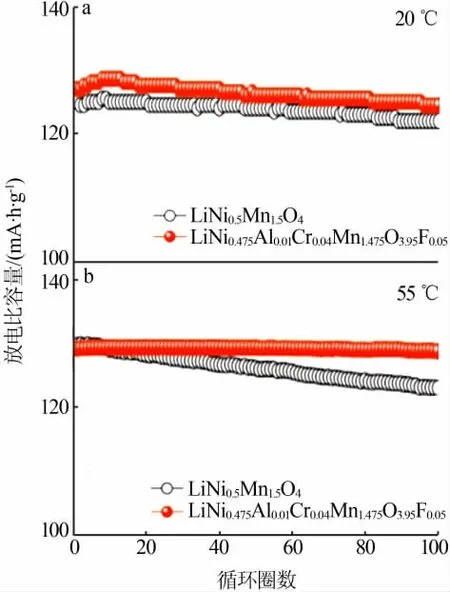
4 总结与展望
