New Approaches for T Cell Immunomodulation
2021-09-10EmanAbdullahBahattabKhalidShahMohannadMokhtarFallatah
Eman Abdullah Bahattab Khalid Shah Mohannad Mokhtar Fallatah



Abstract
Cancer is one of the leading causes of death worldwide. Despite the evolution in cancer treatment, the mortality rate is still high. The astonishing results of immune checkpoint inhibitors in patients bearing solid tumors have encouraged scientists to develop new strategies for cancer immunotherapy to further improve the clinical outcomes. Immunomodulation for cancer therapy is growing rapidly as a promising alternative approach for conventional cancer therapeutics, due to its specificity and efficacy in suppressing growth and metastasis. However, the complexity of the tumor microenvironment and the various mechanisms whereby tumor cells escape the immune response diminishes the efficacy of many cancer immunotherapeutic drugs, especially when the drug is administered as monotherapy. Some of these factors include poor immune recognition of tumor cells, lack of sufficient drug concentration within the tumor site, and suppression of tumor-reactive T cell proliferation and effector functions. This review describes some of the novel immunomodulation strategies that have been recently developed to overcome these limitations and to augment strong antitumor immune response against different tumor types. For example, monoclonal antibodies (mAbs), CAR- T cells, Bi-specific T-cell engagers (BiTE) antibody, oncolytic viruses (OVs), cytokine-antibody fusion protein, genetically engineered mesenchymal stem cells (MSCs), oncolytic virotherapy, gut microbiota modulation and nanoparticles mediated cancer therapy. These novel strategies have been proven to augment strong antitumor immune response against different tumor types, reduced toxicity, as well as generated long-lived memory T cell population that was able to protect the host from tumor relapse. Combination therapies either with conventional or immunotherapy further improved the efficacy of cancer treatment.
Keywords: Immunomodulation; Checkpoint inhibitors; Co-stimulatory receptors; Bispecific antibody;Cytokine/Antibody fusion;Bi-specific T-cell engagers (BiTE) antibody;Oncolytic virus (OVs); Genetically engineered mesenchymal stem cells (MSCs);Nano-based vaccine and Gut microbiota
1. Introduction
The main goal of Immunomodulation strategies is to generate robust antitumor immune response mainly T cells, because they have the ability to specifically bind to and kill tumor cells, their abundance and the ability to differentiate into effector and memory T cell populations for long term protection against tumor relapse [1-3]. Here, we are going to describe some of the novel immunomodulation strategies that have been developed to treat patients with stablished tumor.
2. Checkpoint Inhibitors Restore Effector T Cells Functions and Pproliferation
Immune checkpoints are co-inhibitory molecules including are expressed on immune cells upon activation [4]. They play an important role in inhibiting the immune response to avoid any undesirable collateral damage. However, cancer cells can upregulate co-inhibitory molecules to supress an the activation of naïve and memory T cells to as a mechanism for immune evasion [5].
2.1 Blocking CTLA-4 receptor, where it all started
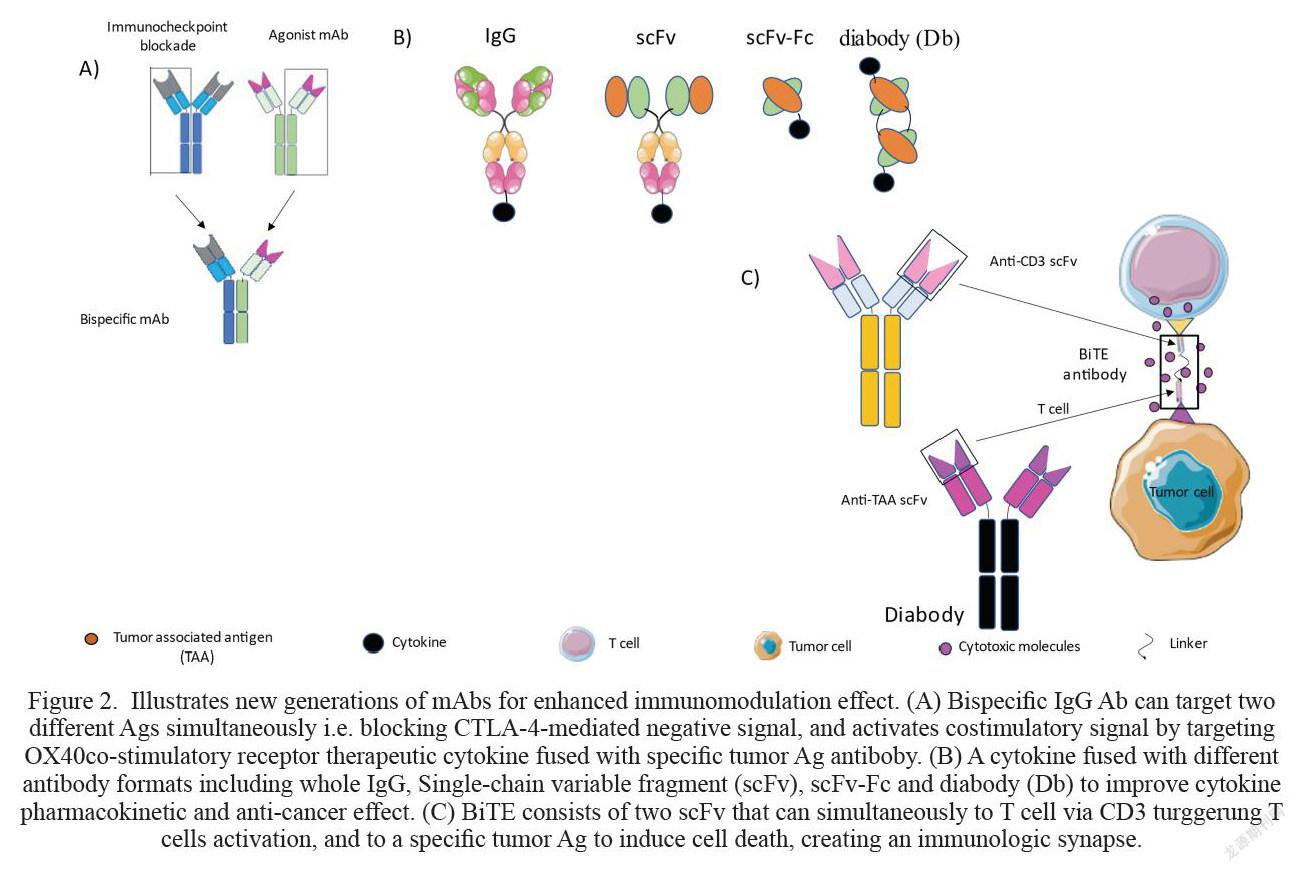
The co-inhibitory receptor cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) is the first co-inhibitory receptor to be discovered [6,7]. CTLA-4 ligation with one of its cognate ligands CD80 or CD86 delivers a negative signal which supress T cell activation. Therefore, blocking antibody (Ab) was developed to interfere with CTLA-4/CD80 or CD86 – mediated co-inhibitory signal to restore cytotoxic T lymphocytes (CTLs) effector function, cytokine secretion and gave rise to an increased number of tumor infiltrating lymphocytes (TILs), thereby, inhibited tumor growth (Fig.1).[8]
2.2. Programmed cell death protein 1 (PD-1)
Programmed cell death protein 1 (PD-1) is coinhibitory receptor that is upregulated transiently on activated T cells and it plays a major role in suppressing T cell proliferation, cytokine production and cytolytic function [11]. Multiple drugs targeting PD-1 or its ligand PD-L1 drugs have been approved by the FDA for the treatment of different tumor types including melanoma, non-small cell lung cancer (NSCLC), kidney, classical Hodgkin lymphoma; and balder cancer [12,13]. Currently, multiple trials examining the efficacy of nivolumab alone or in a form of combined therapy in patients with advanced stage NSCLC [14,15].
The administration of immune check point inhibitor (ICI) as monotherapy is not always sufficient to eliminate the tumor or reduce its growth in patients. Therefore, a combination regimen of anti-CTLA-4 (ipilimumab) and anti-PD-1 (Nivolumab) Abs was proposed to overcome this limitation. The treatment with the combination showed significant improvement in objective response rates (ORR) across tumor and [16,17]. However, cautious approach is warranted given the potential to exacerbate autoimmunity. Furthermore, tumor relapse has been reported in some patients after long follow up, suggesting the emergence of acquired resistance that can be initiated by tumor intrinsic mechanisms such as genetics or epigenetic alterations, and extrinsic mechanisms like immune suppressor cells that act with tumor cells and contribute to tumor progression [5, 18].
Therefore, developing novel therapeutic approaches to treat cancer patients with better efficacy and lower toxicity became mandatory.
Stimulation of Co-stimulatory Receptors as an Alternative Immunomodulation Approach
Targeting costimulatory receptors with agonist mAb is a new immunomodulation approach aiming to generate strong T cell responses against the tumor and protect the host from tumor relapse. Resting naïve CD8+ T cell requires three essential signals to become fully activated and differentiate into effector and memory T cells. The first signal is triggered by the interaction of T cell receptor (TCR) with the foreign antigen (Ag) presented on the major histocompatibility complex (MHC) I by APCs mainly dendritic cells (DCs) [19,20]. The second signal (co-stimulatory signal) is initiated when co-stimulatory receptors on T cells bind to their cognate ligand on DC. These two signals along with proinflammatory cytokine such as interleukin-21 (IL-21) drive naïve CD8+T cell to differentiate into cytotoxic T lymphocytes (CTLs) and memory T cells [19]. The absence of the co-stimulatory signal in many cancer cases can be compensated by targeting co-stimulatory receptors like CD27, OX40, 4-1BB or GITR with agonist mAbs. This approach along with signal 1 triggers CD8+ T cells activation and differentiation into CTLs and long-lived memory CD8+ T cells (Fig.1A). [21,22,23]. Multiple preclinical and clinical studies showed that targeting co-stimulatory receptors with mAb or soluble protein augmented strong immune response, improved tumor cell killing and prolonged mice survival [8, 24-29]. Below we will elaborate on some of the Abs that have been developed to augment strong T cell response or enhance pre-exciting tumor antigen-specific T cells.
3.1 Targeting CD27/CD70 signalling pathway induce strong T cell response
CD27 is expressed on different cell population including NK cells, memory B cells and all T cells subsets [30-33]. This receptor is found on T cells as a transmembrane homodimer in the early stages of the activation. CD27 interaction with CD70 increases the number of effector and memory CD8+ T cell. Treating mice bearing advanced A31 or B-cell lymphoma (BCL1) with Anti-C27 increased CD8+ T cells and improved mice survival compared with the control [34]. Combining anti-CD27 mAb with other immunomodulatory mAb against CD20 Ag (specifically expressed by BCL-1) significantly improved mice survival, such that 100% of the treated mice were tumor free for more than 100 days post treatment [34].
Another study using adoptive CD8+ T cell transfer therapeutic approach in combination with synthetic human gp100 (hgp100) peptide to treat advanced melanoma in mice. Results showed that targeting CD27 co-stimulatory receptor with agonist mAb enhanced antigen-specific CD8+ T cells expansion and effector cytokine production, which was correlated with slower tumor cell growth and longer survival period compared to when mice were injected with hgp100 peptide alone [35].
Currently, the human IgG1κ anti-CD27 antibody varlilumab (CDX-1127) has shown promising signs of immunological and clinical responses in patients with advanced tumor including melanoma and renal cell carcinoma. Patients received escalated doses of varlilumab (0.1, 0.3, 1.0, 3.0, or 10 mg/kg intravenously) with one-month observation, followed by five multidose cycles (one dose per week for 4 weeks). Results were linear and dose dependent with curable adverse effects ranged between grade 1 and 2. One patient experienced a dose-limiting toxicity-grade 3 transient asymptomatic hyponatremia at the 1.0-mg/kg dose level [29]. Eight patients with metastatic renal cell carcinoma (RCC) showed stable disease progression for more than 3 months, including one patient with complete tumor regression for almost 4 years. The biological activity consistent with T cell activation and regulatory T cell depletion at all administered doses. Indicating that varlilumab has biological as well as clinical activity [29]. Wasiuk A. and other have generated 1F5 mouse G1 (promote T cell activation) and 1F5 mouse G2a (depletes Treg suppressor cells) isotype variants of the drug to differentially drive interaction with inhibitory and activating Fcγ receptors (FcγRs), which predictably promoted CD27-mediated agonism or depletion, respectively. There results showed different immunomodulatory effect depending on the isotype tested in human CD27-transgenic mice, both Ab isotypes contributed in the antitumor efficacy of CD27-targeted immunotherapy against BCL1 lymphoma and s.c. injected E.G7 tumor models. Indicating that isotype [36].
3.2 OX40 costimulatory receptor
OX40 is transiently expressed on both CD4+ and CD8+ T cells. Its expression peaks after 48 hours after activation, and down regulates after 72 hours. OX40 interaction with OX40L suppress the function of Treg cells, promotes the expansion of T cell, increase cytokine production and cell migration to tumor site. Moreover, OX40 activation enhances the proliferation of CD4+ T helper (Th) cells and suppress the function of T regulatory (Treg) [37-40].
Early preclinical studies suggested that activating OX40 signalling with agonist Ab or soluble OX40 ligand can induce T cell response and promote tumor cell regression [41]. In a recent study by Peng W et al. showed elevated expression of OX40 on melanoma patients-derive TILs. In vivo, administering agonist Anti-OX40 Ab improved tumor-specific CD8+ T effector function and delayed tumor growth significantly compared with mice treated with control Ab [42]. Moreover, OX40 stimulation successfully suppressed the development of the tumor following tumor challenge, indicating that OX40 agonists can induce the generation of tumor-reactive memory T cells [42].
Recently, a recombinant human OX40L IgG4P Fc fusion protein termed MEDI6383 has been developed to provide agonist activity following engagement of OX40. Exposing previously in vitro activated human T cells to EDI6383 induced 5-fold or higher TNFα and FIN-γ cytokine production compared with T cells treated with the F180A OX40L fusion protein control, increased cell proliferation, and increased the activation markers on both CD4+ and CD8+ T cells [43]. To test the ability of MEDI6383 to mediate tumor clearance in vivo, CD4+ and CD8+ T cells expressing OX40 were mixed with human melanoma cell line A375 and implanted into NOD/SCID mice. Tumor bearing mice then injected with MEDI6383 or F180A as a control. Administering MEDI6383 resulted in a significant tumor growth suppression compared with F180A-treated mice. This effect was mediated by T cells activity as their absence abolished the effect of MEDI6383 [43]. Together, these results indicate that OX40 stimulation has the potential to enhance anti-tumor immunity in human malignancies. Several anti-OX40 agonistic mAbs are currently tested in early phase cancer clinical trials.
Bispecific Abs: Targeting Two Different Ags Simultaneously
The interest to Bispecific IgG1 Abs (BsAb) was significantly increased in the last 10 years. Unlike mAbs which can only bind to one Ag, BsAb can bind to two different Ags simultaneously (Fig. 2A). This feature has led to the generation of number of BsAb for immunomodulation. For example, The ATOR-1015 is a BsAb that was designed to specifically block CTLA-4 signalling pathway and activate OX40 receptor simultaneously (44)]. Injecting mice bearing solid MB49 bladder cancer with multiple injections (248 μg) of ATOR-1015 post tumor inoculation significantly reduced tumor growth and prolonged mice survival, resulting in 7/10 of mice with complete tumor regression for more than 100 days. The anti-tumor effect of ATOR-1015 was superior to anti-OX40 Ab monotherapy, only 2/10 mice were tumor free [44]. Biologically, the BsAb increased CD8+ T cell/Treg ratio in the tumor and upregulated the expression of effector molecules Gransyme B (Grnz B) and CD107a on CD8+ T cells compared to the control. Suggesting the tendency of ATOR-1015 to boost cytotoxic CD8+ T cells activity. Furthermore, ATOR-1015 induced the generation of long-lived memory T cells. This was confirmed by responder mice following the rechallenge with the same tumor MB49. Responsder mice remained tumor free, indicating the formation of memory CD8+ T cells [44]. Catumaxomab is a BsAb that targetsepithelial cell adhesion molecule (EpCAM) and CD3 and is currently in phase II trials for the treatment of patients with gastric cancer. Several advert events associated with Catumaxomab have been reported including nausea, abdominal pain, infection and elevated liver enzymes [45].
Cytokines and Immunomodulation
Cytokines are small glycoproteins that play a major role in regulating the immune response [46]. Despite their tremendous contribution to current clinical cancer research, cytokines-based therapies still have some limitations. For example, cytokines have short half-life and normally act over short distances in a paracrine or autocrine fashion [47,48]. Therefore, cytokine is usually injected in high doses to observe clinical benefits. However, high cytokine doses can lead to severe toxicities such as capillary leak syndrome which can significantly contribute to development of oliguria, ischemia and confusion [49,50]. Furthermore, IL-15 overexpression has been reported to cause leukaemia [47,51]. In addition, in many cases the positive outcomes of cytokine-based therapy were associated with the induction of immunological checkpoints such as IL-10 and transforming growth factor (TGF)-β, myeloid suppressor cells and regulatory T (Treg) cells, all of which contribute to CTLs dysfunction [52]. Therefore, new strategies to overcome these limitations became mandatory.
5.1. Cytokine/Antibody fusion protein overcome side effects of free cytokines
Cytokine/Ab fusion protein strategy has been developed for more efficient cytokine accumulation in the tumor. A wide range of Ab-cytokine fusion formulations been tested over the past decades for their ability to augment strong antitumor immune responses [53]. For example, Interleukin 2 (IL-2) was fused with different antibody formats including whole IgG, Single-chain variable fragment (scFv), scFv-Fc and diabody (Db) (Fig.2B). [53,54]. These constructs improved the pharmacokinetics of the cytokine, and increased its half-life similar to the IgG Ab, and increased their accumulation within the tumor [53,55]. For example, a biodistribution study of immunocompetent mice bearing acute myeloid leukemia (AML) and injected three times (30ug every third day) with F8-IL2 showed selective accumulation of F8-IL-2 in the tumor and inhibited tumor growth significantly for up to day 30 post tumor implantation. The combination of F8-IL2 with chemotherapy resulted in complete tumor regression in 100% of the mice [56]. Notably, the therapeutic effect of F8-IL-2 was abolished when CD8+ or NK cells were depleted, while tumor growth was not affected in the absence of CD4+ T cells were depleted [56]. Indicating that the antitumor immune response is mainly mediated by CD8+ and NK cells populations in mediating tumor growth. The clinical evaluation of the Ab specific to the domain A1 of human tenascin-C (F16-IL-2) also showed promising outcomes, a 51-year-old female patient with refractory AML after two allogeneic stem cell transplantations was treated with F16-IL-2(30 MIU intravenously on day 1, 50 MIU on day 8) and low-dose cytarabine (5 mg twice daily subcutaneously on days 1 to 10). Images obtained from 18-FDG-PET/CT after day 14 following the treatment showed complete tumor regression [56]. Notably, no side effects were reported. On contrary, some clinical symptoms like difficulties in swallowing and restricted head mobility improved after the treatment.
Similar results were obtained for a variety of immunomodulatory cytokines including tumor necrosis factor (TNF), IL-12 and IFN-α2. These cytokines were fused with specific Abs and they were injected intravenously to BALB/c mice with stablished CT26 colon cancer transfected with human carbonic anhydrase IX. The tumor regression rate was variable depending on the cytokine [57]. However, increased proportion of tumour infiltrating Ag-specific CD8+ T cells was observed following Ab-cytokine administration accompanied with prolonged survival rate [57].
In 2015 a novel construct known as PFC-1 and consists of IL-15 cytokine fused with IL-15α, Fc domain and RGD (Arg-Gly-Asp) peptide was developed to enhance the anti-tumor activity of the free cytokine IL-15 [58]. Injecting mice bearing established B16F10 melanoma with PFC-1 reduced tumor cell growth significantly and inhibited tumor metastasis compared to the controlled group which was injected with a vehicle only. Moreover, the ratio of both NK cells and CD8+ T cells in the blood, spleen and in the tumor was significantly higher in PFC-1 treated mice [58].
Cytokines and ICIs is another form of cytokine/Ab fusion protein that have been recently develop for immunotherapy. A fully human IgG1 bifunctional fusion protein designated N-809 encompassing anti-PD-L1 and the IL-15 super agonist fusion complex was designed to potentially mediate Antibody-dependent cellular cytotoxicity (ADCC). N-809 increased the proliferation of human CD4+ and CD8+ T cells, and enhanced their ability to lyse human tumor cells by increasing the expression of perforin and GrnzB [59].
T Cell Redirection Strategies
T cell longevity, functionality and specificity for targeting tumor cell only are key features for successful T cell-mediated therapy. The advancement in knowledge and technology accelerated the development of Chimeric antigen receptors (CARs) T cells for more sufficient tumor cells killing. Redirect TCR by [60, 61]. CARs T cells comprises an extracellular tumor antigen recognition domain fused to the CD3ζ chain and contains one or more co-stimulatory receptors for complete T cell response [62]. Research interest in CARs T cells raised in the last years due to the clinical trials outcome. Two anti-CD19 (CAR) T-cells drugs (Tisagenlecleucel) and (Axicabtagene ciloleucel) received the FDA approval for the treatment of haematological malignancies, acute lymphoblastic leukaemia and large B-cell lymphomas [63,64]. However, cytokine release syndrome (CRS) and neurotoxicity have been repeatedly reported in patients after infusion of CAR-T cells [65,66].
6.1. T cell engagers to tesolve MHC-I Down tegulation on the tumor cells
MHC-I down regulation is one of the immune evasion mechanisms that is used by tumor cells ([67,68]. To overcome this obstacle, bispecific T-cell engager (BiTE) strategy was innovated by infusing dual scFv to kill tumor cell in more fashionable way. One scFv is directed toward the CD3ζ chain for the induction of T cell activation, expansion, and release of cytokine and cytotoxic granules. Surpassing the need of T cell receptor (TCR)/MHC-I ligation which is essential for CD8+ T cells activation. While the other scFv binds to the tumor associated antigen (TAA) and induce direct tumor cell killing (Fig.2C) [69,70]. Blinatumomab is the first FDA approved BiTE consists of two single-chain variable fragments against CD3 and CD19 Ags to treat patients with B-Cell Lymphoma [70,71]. In a clinical trial phase II study, patients received low dose (0,015 milligram (mg)) of Blinatumomab had partial tumor regression, while higher dose (0.06mg) led to complete tumor regression. Interestingly, tumor cells from the bone marrow and liver were also cleared [71]. Treatment-emergent adverse events (TEAEs) have been reported following the administration of blinatumomab, such as cytokine release syndrome (CRS), neurotoxicity, and medication errors [72]. These adverts events are generally consistent with the symptoms associated with relapsed/refractory (R/R) B-cell precursor acute lymphoblastic leukemia (ALL). However, these adverts events are not lethal and can be resolved [72].
Oncolytic viruses (OVs) Specifically Target Cancer Cells and Induce Strong Immune Response Against the Tumor
Oncolytic viruses (OVs) have emerged as powerful cancer therapeutic agents lately due to their specificity and their relatively lower rate of serious adverse effects [73]. OVs use attenuated RNA or DNA viruses to selectively infect and replicate within tumor cells, increasing the availability of TAA as well as increase proinflammatory cytokines release, chemokines, and viral transgene (Fig.3A). These factors as a result initiate potent antitumor immune response or boosting pre-existing native immune response [74,75]. Talimogene laherparepvec (T-VEC) is a modified herpes simplex virus type-1 derived oncolytic virus encoding human cytokine granulocyte-macrophage colony stimulating factor (GM-CSF) to treat advanced melanoma [74,76]. In a randomized phase III study, administering T-VEC intratumorally induced immunologic responses that mediated tumor regression at the primary site as well as metastatic tumor lesions [77,78]. T-VEC was generally well tolerated in patients, the most common reported adverse event was flu-like symptoms [78]. However, oncolytic virotherapy is far from achieving complete tumor regression. Researchers are currently developing variants OVs therapies encoding immunomodulatory agents or costimulatory molecules to be used as monotherapy or in combination with other conventional or immunotherapy approaches to improve its antitumor efficacy.
The Use of Stem Cells for Targeted Therapy
Mesenchymal stromal/stem cells (MSC) are adult multipotent progenitors with fibroblast-like morphology, they have the ability to self-renew and differentiate into a variety of cell types, including adipocytic, osteogenic, chondrogenic, and myogenic lineages [79]. More importantly, MSCs have the ability to home to tumor site, making them a suitable tool for delivering therapeutic agents to the tumor site [80,81]. Because of these favourable features, MSC have been genetically engineered to prolong the half-life of number of immunomodulatory cytokines and reduce their undesired toxicities. Results from a number of in vivo studies to investigate the antitumor immune response of multiple genetically modified MSCs were promising [81-83]. For example, injecting mice bearing established 4T1 breast cancer with MSCs overexpressing mouse IFN-β showed high IFN-β accumulation in the tumour, inhibited primary cancer growth and significantly reduced pulmonary and hepatic metastases compared with plane MSC [83]. Moreover, more splenic CD8+ T cells and mature DCs were observed in MSC-IFN- treated mice compared to the control group. Notably, the percentage of Treg suppressor cells were significantly higher than in MSC-IFN-β treated group [83]. The immunomodulatory effect of MSC-IFN-β was also observed in more aggressive tumor model glioblastoma (GBM) in immunocompetent and immunocompromised (SCID) mice. These mice received MSC-IFN-β or plan MSCs locally after the tumor was resected. Interestingly, MSC-IFN-β had little effect on SCID mice. Mice survival rate was 18 days versus 15 respectively. In contrary, MSC-IFN-β significantly suppressed tumor growth and prolonged immunocompetent mice survival compared to the control group (46 days versus 31), indicating that fully functional immune system is required for full antitumor immune responses [84]. Similar results were obtained in MSCs expressing INF-α in different tumor models ([83,85].
Currently, MSC-IFN-β is in clinical trial phase-I to investigate its efficacy and safety in patients with advanced refractory epithelial ovarian cancer. Results concluded that MSC-IFN-β are incorporated into tumors and locally produce beta-interferon after intraperitoneal (i.p.) injection SCs overexpressing cytokines. Plus, MSC-IFN-β modulated the immune phenotype and facilitated tumor growth control compared to the control. There were no drug-related adverse events [86].
Nanoparticles as an Immunomodulators for Cancer Therapy
Immunizing with tumor antigenic peptides is a simple and attractive form of vaccine strategy designed to augment strong immune repones. Despite their considerable anti-tumor immune effect, peptide-based vaccine is still not sufficient for managing tumor growth [87,88]. Therefore, several adjuvants and peptide delivery strategies are currently under investigation to increase its therapeutic efficacy.
Nanoparticles (NPs) are small materials ranged between 1 and 100nm, and they are currently widely used in the field of medicine for targeted therapy [89]. of Kuai R et al. have developed lipoprotein-mimicking nano discs delivering tumor Ag peptide and CpG Oligodeoxynucleotide (adjuvant) (Fig.3B) for prolonged tumor Ags exposure that could improve the anti-tumor immune response. Results from in vivo studies showed that the administration of Ag/CpG nanodiscs successfully delivered the Ag and the adjuvant to lymphoid organs and exhibited sustained release of Ag/CpG to DCs, resulting in 47-fold increase in CTLs formation compared with soluble vaccine [89]. Furthermore, the combination of Ag/CpG nanodiscs with neo-antigens and checkpoint inhibitors resulted in completed tumor regression in 88% of mice bearing established MC-38 colon carcinoma compared with 25% in the soluble vaccine-treated cohort [89]. Furthermore, Ag/CpG nanodiscs-treated mice remained tumor free following rechallenge with the same tumor with no recorded side effects [89]. Indicating that the vaccine induced the formation of long-lived T cells. These findings represent a new powerful approach for cancer immunotherapy.
The combination of induced immunogenic cell death (ICD) concept with immune adjuvants simultaneously has a considerable interest in immunomodulation. The rationale behind this strategy is to induce tumro cell death via ICD inducers, meanwhile activating an immune response by the adjuvants [90]. In a preclinical study, results showed that injecting mice bearing established A20 lymphoma with low dose (2ug) of doxorubicin (Dox)/CpG NPs led to a significant tumor regression compared to the control group that received injection with PBS only. Combining the treatment with Abs against CTLA-4 and OX40 further enhanced the efficacy of the NPs and induced systemic immune response capable of eradicating distant tumors. The potent antitumor response of the Dox/CpG NPs was also observed in more aggressive tumor models EL4 T cell lymphoma and B16 melanoma [91]. Collectively, Dox/CpG NPs represent an effective and safe tool that could be used as a promising immunomodulatory approach for patients with different types of tumor.
9.1. A STING-activating nanovaccine for cancer immunotherapy.
In addition to the previously mentioned nano vaccines strategies, recent evidence indicates the crucial role of the stimulator of interferon genes (STING) pathway in antitumor immunity [92].
PC7A NP is a mixture of a tumor Ag with synthetic polymeric nanoparticle. This approach generated strong effector T cell responses with no reported with low systemic cytokine expression [92]. The PC7A NP facilitates cytosolic delivery of the tumor Ag to the APCs draining lymph nodes, resulting in improved Ag cross-presentation by the APCs leading to CD8+ T cell stimulation. Meanwhile, type I interferon-stimulated genes are triggered by the activation of STING [92]. In addition, the antitumor effect of PC7A NP-based therapy was notably observed in different tumor types including colon cancer, melanoma and human papilloma virus (HPV) E6/E7 tumor. Tumor size was significantly reduced following the administration of PC7A NP. The combination of PC7A NP with anti-PD-1 blocking Ab led to complete tumor regression without any drug-related side effects in mice initially injected with E6/E7 tumor model [92].
1. Gut Microbiota are Major Immunomodulator
Gut microbiota play a fundamental role in host’s health and well-being. Recent studies described gut microbiota as a major player for modulating cancer immunotherapy toward a particular tumor type [93]. They do so by different mechanisms including by but not limited to metabolism, translocation and immunomodulation [93]. For example, chemotherapy increases small intestine permeability and translocation of selected species of gram-positive bacteria into the secondary lymphoid organs. There, Th-17 cells subsets become activated, thus, facilitating the accumulation of cytotoxic T cells in the tumor site [94]. Moreover, non-responder cancer patients to ICIs is attributed to disruption of gut microbiota [94]. Data collected recently suggested that the efficacy of anti-CTLA-4 Ab depends on certain species of Bacteroides mainly B. thetaiotaomicron orB. fragilis. These findings were supported by the fact that tumor cells did not respond to CTLA-4 blocking Ab in germ free and antibiotic- treated mice. This defect was overcome by transplanting B. fragilis into the recipient mice [95]. Notably, mice received B. fragilis transplantation responded to the treatment similar to normal mice [95]. Likewise, PD-1/PD-L1 pathway blockade seems to rely on specific Bifidobacterium strain. The administration of Bifidobacterium to mice with stablished tumor showed better antitumor immune response to PD-L1 treatment. Moreover, DCs isolated from these mice showed better TAA presentation compared with non-treated group [96]. These findings suggest that gut microbiota modulate cancer immunotherapy. plus, more studies should be conducted to understand the effect of specific microbe strain in the immunomodulation.
Concluding Remarks and Future Perspectives
The clinical success of immune checkpoint inhibitors for the treatment of a growing number of tumor types has accelerated the discovery of new forms of immunotherapy strategies. Yet, the response rate to immunotherapy is still modest. This is largely attributed to the immune suppressive of the tumor microenvironment. Understanding the mechanisms by which tumor cells suppress the immune response can accelerate the discovery of novel immunomodulatory strategies that can elicit a strong antitumor immune response, meanwhile, minimize side effects. Coupling immunomodulation with optimal drug delivery system seems to be compulsory for generating better clinical outcomes, also to decrease any undesirable toxicities. Personalized therapy will be the future of immunotherapy, designing combinational therapy might be the most beneficial option to treat patients with advanced malignancies.
Acknowledgment
We would like to thank all Joint Centers of Excellence Program (JCEP) PIs and members for supporting this project.
References
[1]. Walsh SR, Simovic B, Chen L, Bastin D, Nguyen A, Stephenson K, et al. Endogenous T cells prevent tumor immune escape following adoptive T cell therapy. J Clin Invest. 2019;129(12):5400-10.
[2]. Li D, Li X, Zhou W-L, Huang Y, Liang X, Jiang L, et al. Genetically engineered T cells for cancer immunotherapy. Signal Transduction and Targeted Therapy. 2019;4(1):35.
[3]. Durgeau A, Virk Y, Corgnac S, Mami-Chouaib F. Recent Advances in Targeting CD8 T-Cell Immunity for More Effective Cancer Immunotherapy. Frontiers in Immunology. 2018;9(14).
[4]. Dyck L, Mills KHG. Immune checkpoints and their inhibition in cancer and infectious diseases. Eur J Immunol. 2017;47(5):765-79.
[5]. Donini C, D'Ambrosio L, Grignani G, Aglietta M, Sangiolo D. Next generation immune-checkpoints for cancer therapy. J Thorac Dis. 2018;10(Suppl 13):S1581-S601.
[6]. Velcheti V, Schalper K. Basic Overview of Current Immunotherapy Approaches in Cancer. Am Soc Clin Oncol Educ Book. 2016;35:298-308.
[7]. Gun SY, Lee SWL, Sieow JL, Wong SC. Targeting immune cells for cancer therapy. Redox Biol. 2019;25:101174-.
[8]. Wu L, Yun Z, Tagawa T, Rey-McIntyre K, de Perrot M. CTLA-4 Blockade Expands Infiltrating T Cells and Inhibits Cancer Cell Repopulation during the Intervals of Chemotherapy in Murine Mesothelioma. Molecular Cancer Therapeutics. 2012;11(8):1809-19.
[9]. Tarhini A. Immune-mediated adverse events associated with ipilimumab ctla-4 blockade therapy: the underlying mechanisms and clinical management. Scientifica (Cairo). 2013;2013:857519-.
[10]. Zhao Y, Yang W, Huang Y, Cui R, Li X, Li B. Evolving Roles for Targeting CTLA-4 in Cancer Immunotherapy. Cell Physiol Biochem. 2018;47(2):721-34.
[11]. Riley JL. PD-1 signaling in primary T cells. Immunological reviews. 2009;229(1):114-25.
[12]. Wu X, Gu Z, Chen Y, Chen B, Chen W, Weng L, et al. Application of PD-1 Blockade in Cancer Immunotherapy. Comput Struct Biotechnol J. 2019;17:661-74.
[13].Lee HT, Lee SH, Heo YS. Molecular Interactions of Antibody Drugs Targeting PD-1, PD-L1, and CTLA-4 in Immuno-Oncology. Molecules. 2019;24(6).
[14]. Sundar R, Cho B-C, Brahmer JR, Soo RA. Nivolumab in NSCLC: latest evidence and clinical potential. Ther Adv Med Oncol. 2015;7(2):85-96.
[15]. Chae YK, Arya A, Iams W, Cruz MR, Chandra S, Choi J, et al. Current landscape and future of dual anti-CTLA4 and PD-1/PD-L1 blockade immunotherapy in cancer; lessons learned from clinical trials with melanoma and non-small cell lung cancer (NSCLC). Journal for ImmunoTherapy of Cancer. 2018;6(1):39.
[16]. Rotte A. Combination of CTLA-4 and PD-1 blockers for treatment of cancer. Journal of Experimental & Clinical Cancer Research. 2019;38(1):255.
[17]. Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Rutkowski P, Lao CD, et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N Engl J Med. 2019;381(16):1535-46.
[18]. Fares CM, Van Allen EM, Drake CG, Allison JP, Hu-Lieskovan S. Mechanisms of Resistance to Immune Checkpoint Blockade: Why Does Checkpoint Inhibitor Immunotherapy Not Work for All Patients? Am Soc Clin Oncol Educ Book. 2019;39:147-64.
[19]. Heath WR, Belz GT, Behrens GM, Smith CM, Forehan SP, Parish IA, et al. Cross-presentation, dendritic cell subsets, and the generation of immunity to cellular antigens. Immunol Rev. 2004;199:9-26.
[20]. Bousso P. T-cell activation by dendritic cells in the lymph node: lessons from the movies. Nat Rev Immunol. 2008;8(9):675-84.
[21]. Ward-Kavanagh LK, Lin WW, Sedy JR, Ware CF. The TNF Receptor Superfamily in Co-stimulating and Co-inhibitory Responses. Immunity. 2016;44(5):1005-19.
[22]. Linsley PS, Ledbetter JA. The role of the CD28 receptor during T cell responses to antigen. Annu Rev Immunol. 1993;11:191-212.
[23]. Attanasio J, Wherry EJ. Costimulatory and Coinhibitory Receptor Pathways in Infectious Disease. Immunity. 2016;44(5):1052-68.
[24]. Buchan SL, Fallatah M, Thirdborough SM, Taraban VY, Rogel A, Thomas LJ, et al. PD-1 Blockade and CD27 Stimulation Activate Distinct Transcriptional Programs That Synergize for CD8(+) T-Cell-Driven Antitumor Immunity. Clinical cancer research : an official journal of the American Association for Cancer Research. 2018;24(10):2383-94.
[25]. Huang J, Jochems C, Anderson AM, Talaie T, Jales A, Madan RA, et al. Soluble CD27-pool in humans may contribute to T cell activation and tumor immunity. Journal of immunology (Baltimore, Md : 1950). 2013;190(12):6250-8.
[26]. Buchan SL, Dou L, Remer M, Booth SG, Dunn SN, Lai C, et al. Antibodies to Costimulatory Receptor 4-1BB Enhance Anti-tumor Immunity via T Regulatory Cell Depletion and Promotion of CD8 T Cell Effector Function. Immunity. 2018;49(5):958-70.e7.
[27]. Pan PY, Zang Y, Weber K, Meseck ML, Chen SH. OX40 ligation enhances primary and memory cytotoxic T lymphocyte responses in an immunotherapy for hepatic colon metastases. Mol Ther. 2002;6(4):528-36.
[28]. Reiss KA, Forde PM, Brahmer JR. Harnessing the power of the immune system via blockade of PD-1 and PD-L1: a promising new anticancer strategy. Immunotherapy. 2014;6(4):459-75.
[29]. Burris HA, Infante JR, Ansell SM, Nemunaitis JJ, Weiss GR, Villalobos VM, et al. Safety and Activity of Varlilumab, a Novel and First-in-Class Agonist Anti-CD27 Antibody, in Patients With Advanced Solid Tumors. J Clin Oncol. 2017;35(18):2028-36.
[30]. Hintzen RQ, de Jong R, Lens SM, Brouwer M, Baars P, van Lier RA. Regulation of CD27 expression on subsets of mature T-lymphocytes. The Journal of Immunology. 1993;151(5):2426-35.
[31]. Hayakawa Y, Smyth MJ. CD27 Dissects Mature NK Cells into Two Subsets with Distinct Responsiveness and Migratory Capacity. The Journal of Immunology. 2006;176(3):1517-24.
[32]. Prasad KV, Ao Z, Yoon Y, Wu MX, Rizk M, Jacquot S, et al. CD27, a member of the tumor necrosis factor receptor family, induces apoptosis and binds to Siva, a proapoptotic protein. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(12):6346-51.
[33]. Wu YC, Kipling D, Dunn-Walters DK. The relationship between CD27 negative and positive B cell populations in human peripheral blood. Front Immunol. 2011;2:81.
[34].Turaj AH, Hussain K, Cox KL, Rose-Zerilli MJJ, Testa J, Dahal LN, et al. Antibody Tumor Targeting Is Enhanced by CD27 Agonists through Myeloid Recruitment. Cancer Cell. 2017;32(6):777-91.e6.
[35].Buchan SL, Fallatah M, Thirdborough SM, Taraban VY, Rogel A, Thomas LJ, et al. PD-1 Blockade and CD27 Stimulation Activate Distinct Transcriptional Programs That Synergize for CD8(+) T-Cell-Driven Antitumor Immunity. Clin Cancer Res. 2018;24(10):2383-94.
[36].Wasiuk A, Testa J, Weidlick J, Sisson C, Vitale L, Widger J, et al. CD27-Mediated Regulatory T Cell Depletion and Effector T Cell Costimulation Both Contribute to Antitumor Efficacy. J Immunol. 2017;199(12):4110-23.
[37].Aspeslagh S, Postel-Vinay S, Rusakiewicz S, Soria JC, Zitvogel L, Marabelle A. Rationale for anti-OX40 cancer immunotherapy. Eur J Cancer. 2016;52:50-66.
[38].Voo KS, Bover L, Harline ML, Vien LT, Facchinetti V, Arima K, et al. Antibodies targeting human OX40 expand effector T cells and block inducible and natural regulatory T cell function. J Immunol. 2013;191(7):3641-50.
[39].Polesso F, Sarker M, Weinberg AD, Murray SE, Moran AE. OX40 Agonist Tumor Immunotherapy Does Not Impact Regulatory T Cell Suppressive Function. The Journal of Immunology. 2019:ji1900696.
[40].Linch SN, McNamara MJ, Redmond WL. OX40 Agonists and Combination Immunotherapy: Putting the Pedal to the Metal. Frontiers in Oncology. 2015;5(34).
[41].Weinberg AD, Rivera MM, Prell R, Morris A, Ramstad T, Vetto JT, et al. Engagement of the OX-40 receptor in vivo enhances antitumor immunity. J Immunol. 2000;164(4):2160-9.
[42].Peng W, Williams LJ, Xu C, Melendez B, McKenzie JA, Chen Y, et al. Anti-OX40 Antibody Directly Enhances The Function of Tumor-Reactive CD8(+) T Cells and Synergizes with PI3Kbeta Inhibition in PTEN Loss Melanoma. Clin Cancer Res. 2019;25(21):6406-16.
[43].Oberst MD, Auge C, Morris C, Kentner S, Mulgrew K, McGlinchey K, et al. Potent Immune Modulation by MEDI6383, an Engineered Human OX40 Ligand IgG4P Fc Fusion Protein. Mol Cancer Ther. 2018;17(5):1024-38.
[44].Kvarnhammar AM, Veitonmaki N, Hagerbrand K, Dahlman A, Smith KE, Fritzell S, et al. The CTLA-4 x OX40 bispecific antibody ATOR-1015 induces anti-tumor effects through tumor-directed immune activation. J Immunother Cancer. 2019;7(1):103.
[45].Knödler M, Körfer J, Kunzmann V, Trojan J, Daum S, Schenk M, et al. Randomised phase II trial to investigate catumaxomab (anti-EpCAM × anti-CD3) for treatment of peritoneal carcinomatosis in patients with gastric cancer. British Journal of Cancer. 2018;119(3):296-302.
[46].Oberholzer A, Oberholzer C, Moldawer LL. Cytokine signaling--regulation of the immune response in normal and critically ill states. Crit Care Med. 2000;28(4 Suppl):N3-12.
[47].Robinson TO, Schluns KS. The potential and promise of IL-15 in immuno-oncogenic therapies. Immunol Lett. 2017;190:159-68.
[48].Hu Q, Ye X, Qu X, Cui D, Zhang L, Xu Z, et al. Discovery of a novel IL-15 based protein with improved developability and efficacy for cancer immunotherapy. Scientific Reports. 2018;8(1):7675.
[49].Schwartz RN, Stover L, Dutcher JP. Managing toxicities of high-dose interleukin-2. Oncology (Williston Park). 2002;16(11 Suppl 13):11-20.
[50].Panelli MC, White R, Foster M, Martin B, Wang E, Smith K, et al. Forecasting the cytokine storm following systemic interleukin (IL)-2 administration. J Transl Med. 2004;2(1):17-.
[51].Fehniger TA, Suzuki K, Ponnappan A, VanDeusen JB, Cooper MA, Florea SM, et al. Fatal leukemia in interleukin 15 transgenic mice follows early expansions in natural killer and memory phenotype CD8+ T cells. J Exp Med. 2001;193(2):219-31.
[52].Taylor A, Verhagen J, Blaser K, Akdis M, Akdis CA. Mechanisms of immune suppression by interleukin-10 and transforming growth factor-beta: the role of T regulatory cells. Immunology. 2006;117(4):433-42.
[53].Neri D. Antibody-Cytokine Fusions: Versatile Products for the Modulation of Anticancer Immunity. Cancer Immunol Res. 2019;7(3):348-54.
[54].Hutmacher C, Neri D. Antibody-cytokine fusion proteins: Biopharmaceuticals with immunomodulatory properties for cancer therapy. Adv Drug Deliv Rev. 2019;141:67-91.
[55].Gillies SD, Lo KM, Burger C, Lan Y, Dahl T, Wong WK. Improved circulating half-life and efficacy of an antibody-interleukin 2 immunocytokine based on reduced intracellular proteolysis. Clin Cancer Res. 2002;8(1):210-6.
[56].Gutbrodt KL, Schliemann C, Giovannoni L, Frey K, Pabst T, Klapper W, et al. Antibody-Based Delivery of Interleukin-2 to Neovasculature Has Potent Activity Against Acute Myeloid Leukemia. Science Translational Medicine. 2013;5(201):201ra118-201ra118.
[57].Ziffels B, Stringhini M, Probst P, Fugmann T, Sturm T, Neri D. Antibody-Based Delivery of Cytokine Payloads to Carbonic Anhydrase IX Leads to Cancer Cures in Immunocompetent Tumor-Bearing Mice. Mol Cancer Ther. 2019;18(9):1544-54.
[58].Chen S, Huang Q, Liu J, Xing J, Zhang N, Liu Y, et al. A targeted IL-15 fusion protein with potent anti-tumor activity. Cancer Biol Ther. 2015;16(9):1415-21.
[59].Jochems C, Tritsch SR, Knudson KM, Gameiro SR, Rumfield CS, Pellom ST, et al. The multi-functionality of N-809, a novel fusion protein encompassing anti-PD-L1 and the IL-15 superagonist fusion complex. Oncoimmunology. 2019;8(2):e1532764.
[60].Srivastava S, Riddell SR. Engineering CAR-T cells: Design concepts. Trends Immunol. 2015;36(8):494-502.
[61].Guedan S, Calderon H, Posey AD, Jr., Maus MV. Engineering and Design of Chimeric Antigen Receptors. Mol Ther Methods Clin Dev. 2019;12:145-56.
[62].Feins S, Kong W, Williams EF, Milone MC, Fraietta JA. An introduction to chimeric antigen receptor (CAR) T-cell immunotherapy for human cancer. Am J Hematol. 2019;94(S1):S3-s9.
[63].Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. New England Journal of Medicine. 2017;377(26):2531-44.
[64].Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. New England Journal of Medicine. 2018;378(5):439-48.
[65].Frey N, Porter D. Cytokine Release Syndrome with Chimeric Antigen Receptor T Cell Therapy. Biol Blood Marrow Transplant. 2019;25(4):e123-e7.
[66].Hunter BD, Jacobson CA. CAR T-Cell Associated Neurotoxicity: Mechanisms, Clinicopathologic Correlates, and Future Directions. J Natl Cancer Inst. 2019;111(7):646-54.
[67].Rodríguez JA. HLA-mediated tumor escape mechanisms that may impair immunotherapy clinical outcomes via T-cell activation. Oncol Lett. 2017;14(4):4415-27.
[68].Garrido F, Aptsiauri N, Doorduijn EM, Garcia Lora AM, van Hall T. The urgent need to recover MHC class I in cancers for effective immunotherapy. Curr Opin Immunol. 2016;39:44-51.
[69].Zhukovsky EA, Morse RJ, Maus MV. Bispecific antibodies and CARs: generalized immunotherapeutics harnessing T cell redirection. Curr Opin Immunol. 2016;40:24-35.
[70].Smits NC, Sentman CL. Bispecific T-Cell Engagers (BiTEs) as Treatment of B-Cell Lymphoma. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2016;34(10):1131-3.
[71].Bargou R, Leo E, Zugmaier G, Klinger M, Goebeler M, Knop S, et al. Tumor regression in cancer patients by very low doses of a T cell-engaging antibody. Science. 2008;321(5891):974-7.
[72].Stein A, Franklin JL, Chia VM, Arrindell D, Kormany W, Wright J, et al. Benefit-Risk Assessment of Blinatumomab in the Treatment of Relapsed/Refractory B-Cell Precursor Acute Lymphoblastic Leukemia. Drug Saf. 2019;42(5):587-601.
[73].Orange M, Reuter U, Hobohm U. Coley's Lessons Remembered: Augmenting Mistletoe Therapy. Integr Cancer Ther. 2016;15(4):502-11.
[74].Sivanandam V, LaRocca CJ, Chen NG, Fong Y, Warner SG. Oncolytic Viruses and Immune Checkpoint Inhibition: The Best of Both Worlds. Mol Ther Oncolytics. 2019;13:93-106.
[75].Schirrmacher V. From chemotherapy to biological therapy: A review of novel concepts to reduce the side effects of systemic cancer treatment (Review). Int J Oncol. 2019;54(2):407-19.
[76].Harrington KJ, Puzanov I, Hecht JR, Hodi FS, Szabo Z, Murugappan S, et al. Clinical development of talimogene laherparepvec (T-VEC): a modified herpes simplex virus type-1-derived oncolytic immunotherapy. Expert Rev Anticancer Ther. 2015;15(12):1389-403.
[77].Russell L, Peng KW, Russell SJ, Diaz RM. Oncolytic Viruses: Priming Time for Cancer Immunotherapy. BioDrugs. 2019;33(5):485-501.
[78].Kaufman HL, Kim DW, DeRaffele G, Mitcham J, Coffin RS, Kim-Schulze S. Local and distant immunity induced by intralesional vaccination with an oncolytic herpes virus encoding GM-CSF in patients with stage IIIc and IV melanoma. Ann Surg Oncol. 2010;17(3):718-30.
[79].D'Souza N, Burns JS, Grisendi G, Candini O, Veronesi E, Piccinno S, et al. MSC and Tumors: Homing, Differentiation, and Secretion Influence Therapeutic Potential. Adv Biochem Eng Biotechnol. 2013;130:209-66.
[80].Reagan MR, Kaplan DL. Concise review: Mesenchymal stem cell tumor-homing: detection methods in disease model systems. Stem Cells. 2011;29(6):920-7.
[81].Shah K. Mesenchymal stem cells engineered for cancer therapy. Adv Drug Deliv Rev. 2012;64(8):739-48.
[82].Kidd S, Caldwell L, Dietrich M, Samudio I, Spaeth EL, Watson K, et al. Mesenchymal stromal cells alone or expressing interferon-beta suppress pancreatic tumors in vivo, an effect countered by anti-inflammatory treatment. Cytotherapy. 2010;12(5):615-25.
[83].Ren C, Kumar S, Chanda D, Kallman L, Chen J, Mountz JD, et al. Cancer gene therapy using mesenchymal stem cells expressing interferon-beta in a mouse prostate cancer lung metastasis model. Gene Ther. 2008;15(21):1446-53.
[84].Choi SH, Stuckey DW, Pignatta S, Reinshagen C, Khalsa JK, Roozendaal N, et al. Tumor Resection Recruits Effector T Cells and Boosts Therapeutic Efficacy of Encapsulated Stem Cells Expressing IFNbeta in Glioblastomas. Clin Cancer Res. 2017;23(22):7047-58.
[85].Xu C, Lin L, Cao G, Chen Q, Shou P, Huang Y, et al. Interferon-alpha-secreting mesenchymal stem cells exert potent antitumor effect in vivo. Oncogene. 2014;33(42):5047-52.
[86].Andreeff M, Marini FC, Westin SN, Coleman RL, Thall PF, Aljahdami V, et al. Abstract 75: A phase I trial of mesenchymal stem cells transfected with a plasmid secreting interferon beta in advanced ovarian cancer. Cancer Research. 2018;78(13 Supplement):75-.
[87].Guo Y, Lei K, Tang L. Neoantigen Vaccine Delivery for Personalized Anticancer Immunotherapy. Frontiers in Immunology. 2018;9(1499).
[88].Bezu L, Kepp O, Cerrato G, Pol J, Fucikova J, Spisek R, et al. Trial watch: Peptide-based vaccines in anticancer therapy. Oncoimmunology. 2018;7(12):e1511506-e.
[89].Kuai R, Ochyl LJ, Bahjat KS, Schwendeman A, Moon JJ. Designer vaccine nanodiscs for personalized cancer immunotherapy. Nat Mater. 2017;16(4):489-96.
[90].Yoon HY, Selvan ST, Yang Y, Kim MJ, Yi DK, Kwon IC, et al. Engineering nanoparticle strategies for effective cancer immunotherapy. Biomaterials. 2018;178:597-607.
[91].Makkouk A, Joshi VB, Wongrakpanich A, Lemke CD, Gross BP, Salem AK, et al. Biodegradable microparticles loaded with doxorubicin and CpG ODN for in situ immunization against cancer. Aaps j. 2015;17(1):184-93.
[92].Luo M, Liu Z, Zhang X, Han C, Samandi LZ, Dong C, et al. Synergistic STING activation by PC7A nanovaccine and ionizing radiation improves cancer immunotherapy. J Control Release. 2019;300:154-60.
[93].Fan Y, Pedersen O. Gut microbiota in human metabolic health and disease. Nature Reviews Microbiology. 2021;19(1):55-71.
[94].Viaud S, Saccheri F, Mignot G, Yamazaki T, Daillère R, Hannani D, et al. The intestinal microbiota modulates the anticancer immune effects of cyclophosphamide. Science. 2013;342(6161):971-6.
[95].Vétizou M, Pitt JM, Daillère R, Lepage P, Waldschmitt N, Flament C, et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science. 2015;350(6264):1079-84.
[96].Sivan A, Corrales L, Hubert N, Williams JB, Aquino-Michaels K, Earley ZM, et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science. 2015;350(6264):1084-9.
杂志排行

Trends in Oncology的其它文章
- Prognostic Biomarkers in Patients with Renal Cell Carcinoma: Where are We Going from Here?
- Does Liquorice Root, Hawthorn Fruit and Chinese Plum Tea Improve Quality of Life in Head and Neck Cancer Patients after Primary or Adjuvant Radiotherapy?
- Gut Microbiota: The Servantof Human Being and the Accessary of Tumorigenesis
- PVSRIPO, APotential New Tool in Our Fight against GBM