定点偶联技术在抗体药物偶联物中的应用
2021-05-11李明莹汪琳马宁宁
李明莹,汪琳,马宁宁*
(1.沈阳药科大学无涯创新学院,辽宁 沈阳 110016;2.沈阳药科大学生命科学与生物制药学院,辽宁 沈阳 110016)
1 引言
化疗是肿瘤前期治疗的主要手段,化学药物在杀伤肿瘤细胞的同时,对正常细胞也会造成杀伤作用。为了提高药物的肿瘤靶向性,抗体药物偶联物(ADC)应运而生。ADC 利用连接子将单克隆抗体(以下简称单抗)与小分子药物连接(见图1),利用单抗的特异性,ADC 可准确地作用于靶点,降低药物对正常细胞的毒副作用。ADC 兼具抗体药物的特异性、稳定性和小分子毒素对肿瘤细胞的药效学特性,是目前抗肿瘤药物研究的热点方向之一[1-2]。至今共有10 款ADC 获美国FDA 批准上市(见表1)。
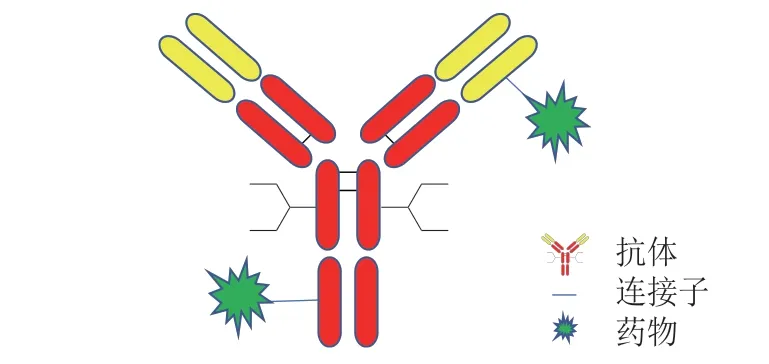
图1 抗体药物偶联物结构示意图Figure 1 Structural diagram of antibody-drug conjugates
传统的ADC 利用抗体赖氨酸的氨基或打开链间二硫键获得的半胱氨酸的巯基进行偶联,赖氨酸的氨基与活化的羧酸酯连接子通过酰胺键连接,半胱氨酸的巯基与马来酰亚胺基团反应[3-4],其偶联示意图如图2 所示。一个抗体分子包含了80 ~ 90 个赖氨酸,偶联可能会发生在将近40 个不同赖氨酸残基上,打开链间二硫键会得到多个半胱氨酸残基,同时破坏了抗体分子的完整性,因此传统的ADC 是高度异质混合物,其均一性差,稳定性低,影响药效及治疗窗[5]。定点偶联技术可以实现抗体与小分子毒素定点、定量偶联,通过该技术获得的ADC具有合适的药抗比(drug antibody ratio,DAR),均一性高,稳定性好,批次间重现性高,具有更好的活性和药动学特性,同时也更适用于ADC 的大规模生产[6]。本文将对定点偶联技术在ADC 开发中的应用进行综述。
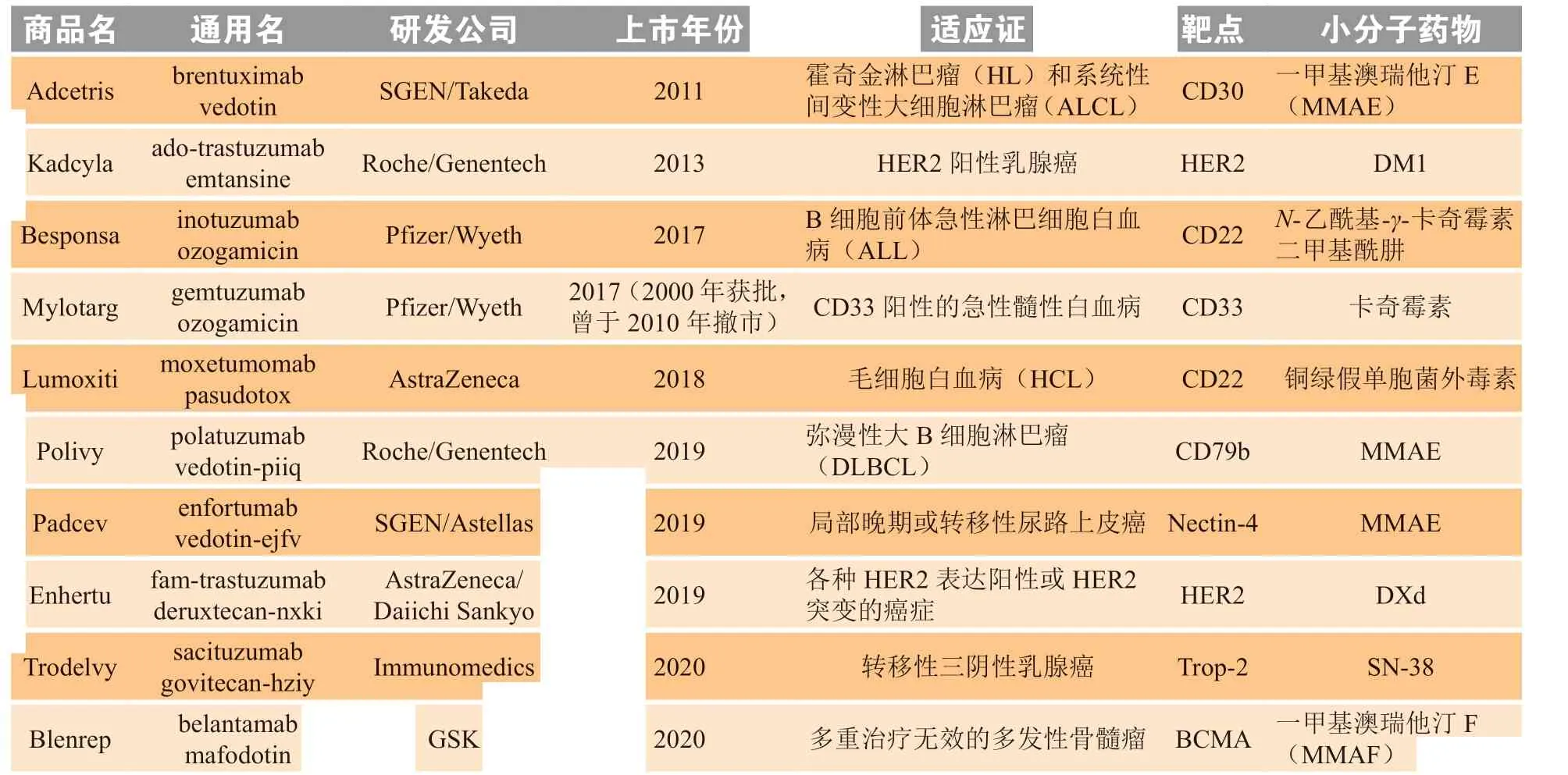
表1 已获FDA 批准上市的抗体药物偶联物Table 1 Antibody-drug conjugates approved by FDA for marketing
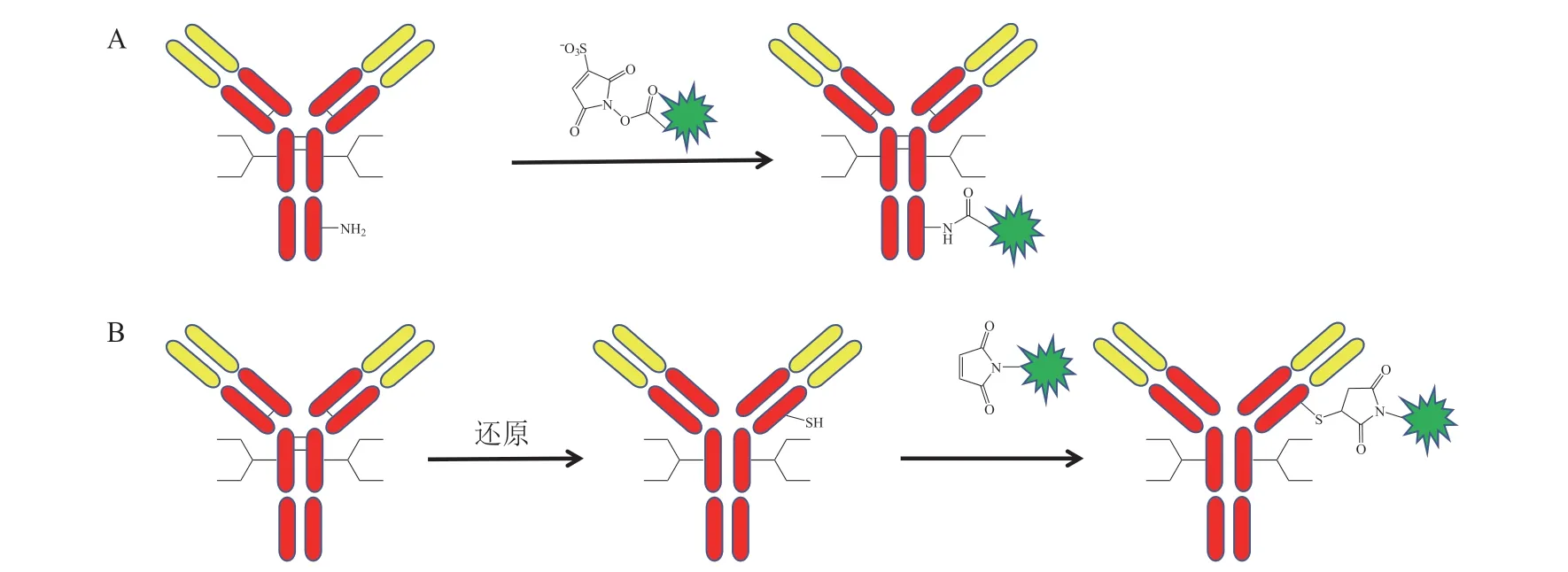
图2 非定点偶联示意图Figure 2 Schematic diagram of non-site-specific conjugation
2 引入非天然氨基酸的定点偶联
天然氨基酸中可用于偶联的氨基酸只有赖氨酸和半胱氨酸,非天然氨基酸可被构建到重组蛋白中,获得可与小分子药物发生化学反应的残基侧链。因此非天然氨基酸为ADC 的开发提供了一个新的技术手段。通过非天然氨基酸可以实现抗体-药物位点特异性偶联。蛋白质在核糖体上的合成通过tRNA反密码子与mRNA 密码子识别来进行[7]。Ambrx 公司引入可特异性识别非天然氨基酸的tRNA 和与之相对应的氨酰tRNA 合成酶,在氨酰tRNA 合成酶的作用下,tRNA 与对应的非天然氨基酸结合形成氨酰tRNA,再通过其反密码子与mRNA 上的密码子互补,使非天然氨基酸被整合到多肽链中,合成含非天然氨基酸的重组抗体[8]。
引入的非天然氨基酸通常为乙酰苯丙氨酸(1)、叠氮基甲基-L-苯基丙氨酸(2)和叠氮赖氨酸(3)。非天然氨基酸上带有的酮基、叠氮基官能团可与药物连接子发生化学反应,获得DAR 均一的ADC。酮基可与羟胺基团形成肟键(见图3A),叠氮基团可与炔基在铜的催化下形成1,2,3-三唑的环加成反应(见图3B),叠氮基团还可以在没有铜催化的情况下与环辛炔结合,发生叠氮-辛炔环加成反应(见图3C)[9-12]。引入非天然氨基酸的抗体与药物连接子可定点、定量偶联,获得DAR 均一、药效高、稳定性好、安全性高的ADC,但也存在抗体表达困难,易产生免疫原性的弊端。Ambrx 公司的ARX788 是首个利用非天然氨基酸开发的抗体偶联药物,目前处于临床研究阶段。
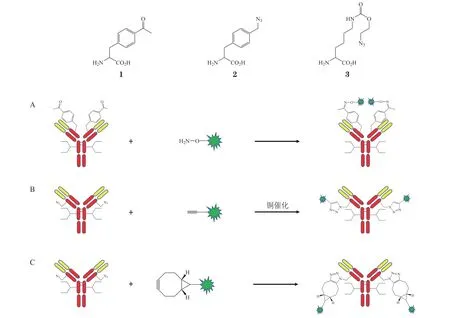
图3 引入非天然氨基酸的定点偶联Figure 3 Site-specific conjugation with insertion of non-natural amino acids
3 酶法偶联
酶具有高特异性和高效性,现也应用于ADC的开发,抗体和小分子药物可以利用酶法实现定点偶联。酶法偶联具备定点偶联的优势,但也存在因引入额外序列而造成免疫原性的潜在弊端。以下将对4 种酶在ADC 开发中的应用进行介绍。
3.1 谷氨酰胺转移酶
谷氨酰胺转移酶(TGase)催化谷氨酰胺与赖氨酸及其衍生物反应,通过TGase 也可以实现抗体和药物定点偶联。野生型TGase 将药物连接子的胺转移到去糖基化的抗体重链Q295 中(见图4A)[13]。Innate Pharm 对抗体重链连接糖链的N297 进行突变,再通过TGase 实现抗体药物偶联(见图4B)[14]。辉瑞提出在抗体上插入谷氨酰胺标记的LLQGA 五肽序列,通过TGase 特异性识别LLQGA 五肽序列中的谷氨酰胺(见图4C),将药物与其进行偶联[15]。
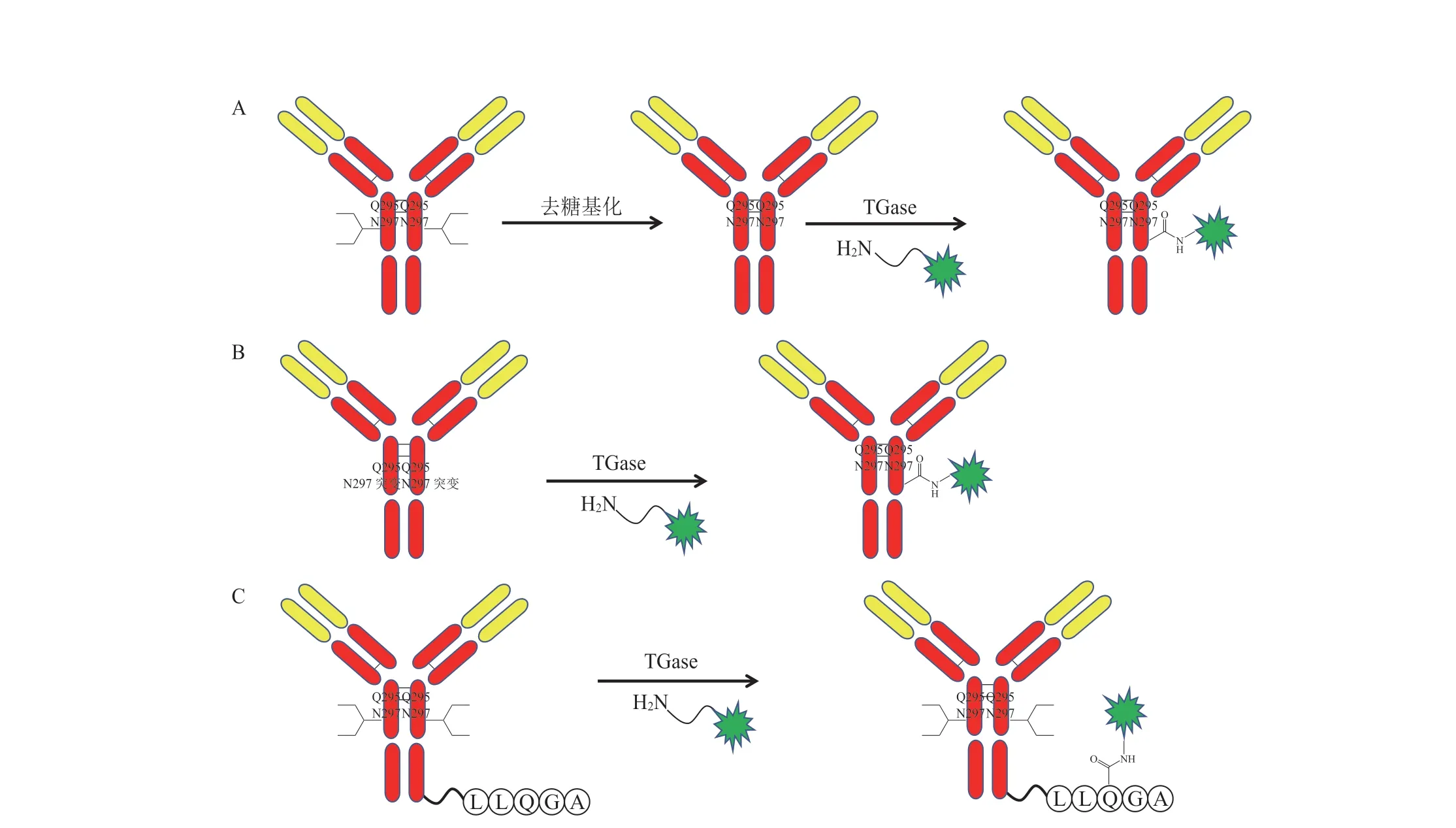
图4 应用谷氨酰胺转移酶进行定点偶联Figure 4 Site-specific conjugation using transglutaminase
3.2 分选酶
分选酶普遍存在于革兰阳性菌中,具有转肽催化作用,可特异性识别LPXTG 序列,并打开苏氨酸和甘氨酸之间的肽键,插入重复的甘氨酸序列。通过这项技术,Beerli 等[16]在抗体的重链和轻链的C 末端插入了LPETG 序列,与带有甘氨酸链的MMAE 偶联(见图5),从而实现抗体药物定点偶联。
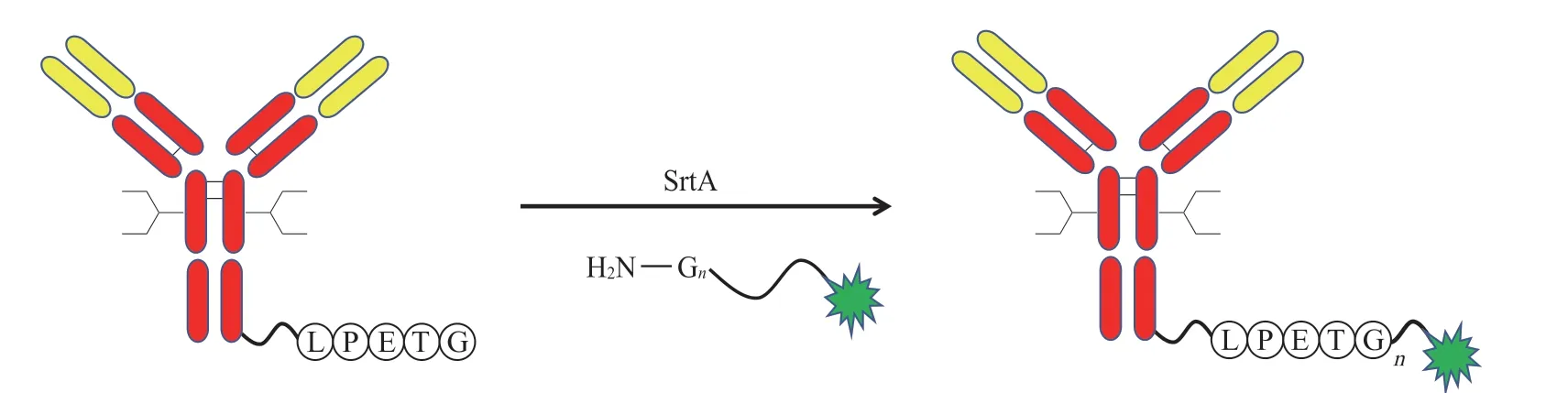
图5 应用分选酶进行定点偶联Figure 5 Site-specific conjugation using sortase
3.3 甲酰甘氨酸生成酶
甲酰甘氨酸生成酶(FGE)可特异性识别CXPXR 五肽序列,将半胱氨酸的残基替换成醛基,与二甲基化的2-(联氨甲基)-3-吲哚发生反应,在接近中性的pH 值下,通过四氢异喹啉合成(HIPS)反应形成稳定的碳碳键(见图6)[17]。Redwood Bioscience 公司的专利SMARTag™技术即采用FGE来实现抗体与药物的定点偶联[18]。
3.4 类异戊二烯转移酶
LegoChemBio 利用类异戊二烯转移酶技术开发ADC[19]。在抗体C 端插入由几个甘氨酸序列连接的CAAX 序列,利用类异戊二烯转移酶将异戊二烯基连接在CAAX 序列的半胱氨酸残基上,再与连接子发生肟连接反应(见图7),从而实现抗体药物定点偶联[20]。
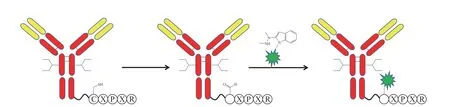
图6 应用甲酰甘氨酸生成酶进行定点偶联Figure 6 Site-specific conjugation using formatylglycine generating enzyme
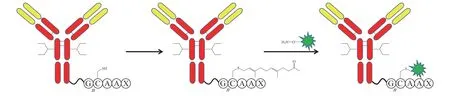
图7 应用类异戊二烯转移酶进行定点偶联Figure 7 Site-specific conjugation using isoprene transferase
4 糖链重塑和糖基偶联
抗体在Fc 段含有糖链,可利用糖链重塑和糖基进行抗体药物定点偶联。研究人员利用β-1,4-半乳糖基转移酶(Gal T)和α-2,6-唾液酸转移酶(Sial T)将半乳糖和唾液酸残基转移到抗体的天然糖基上,然后应用高碘酸钠(NaIO4)氧化半乳糖或唾液酸中的顺乙二醇基团,引入醛基,利用醛与含有肼或伯胺官能团的分子发生反应,进而实现定点偶联,得到DAR 均一的ADC(见图8A)[21-22]。文献报道,还可利用糖基转移酶引入叠氮基,利用叠氮与环辛炔反应形成稳定的ADC(见图8B)[23-24]。
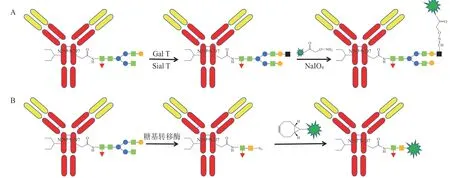
图8 应用糖链重塑和糖基进行定点偶联Figure 8 Site-specific conjugation using saccharide chain remodeling and saccharide
5 基于反应性半胱氨酸的定点偶联
抗体表面不存在可以用于偶联反应的半胱氨酸残基,其均以二硫键的形式存在。例如最常见的IgG1,其含有4 个链间二硫键和12 个链内二硫键,使用传统的偶联方法,要先打开4 个链间二硫键来获得游离巯基,该方法制备的ADC 的DAR 值在0 ~ 8之间,均一性较差。通过基于反应性半胱氨酸的定点偶联可获得DAR 值确定的ADC。
5.1 基于链间二硫键改造的定点偶联
抗体的轻重链由链间二硫键连接,打开抗体的链间二硫键对抗体结构和功能的影响较小,可获得确定的反应性半胱氨酸,因此可通过链间二硫键改造实现抗体药物定点偶联。选择合适的还原剂将抗体的链间二硫键还原,再将连有二磺酸盐(见图9A)或二溴双反应试剂(见图9B)的小分子毒素与还原的抗体反应,从而实现抗体药物定点偶联[25-26]。但利用该方法获得的ADC 存在偶联效率低的弊端。
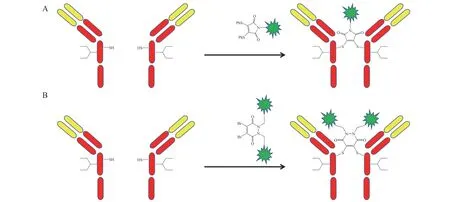
图9 基于链间二硫键改造的定点偶联Figure 9 Site-specific conjugation based on interchain disulfide bond modification
5.2 Thio-Mab 技术
Thio-Mab 这一概念最早由Genentech 提出,是基于噬菌体展示的方法,在抗体的Fab 表面筛选出反应性半胱氨酸的突变位点。Genentech 最终选择曲妥珠单抗的LC-V110 和HC-A114(Kabat 编号)进行半胱氨酸突变,利用三(2-羧乙基)膦(TCEP)或二硫苏糖醇(DTT)只打开抗体链间二硫键,并使突变的半胱氨酸的巯基处于自由态,再应用CuSO4或脱氢抗坏血酸(dhAA)将链间二硫键重新连接,最后利用游离巯基与药物连接子反应,实现抗体药物定点偶联(见图10)[27]。
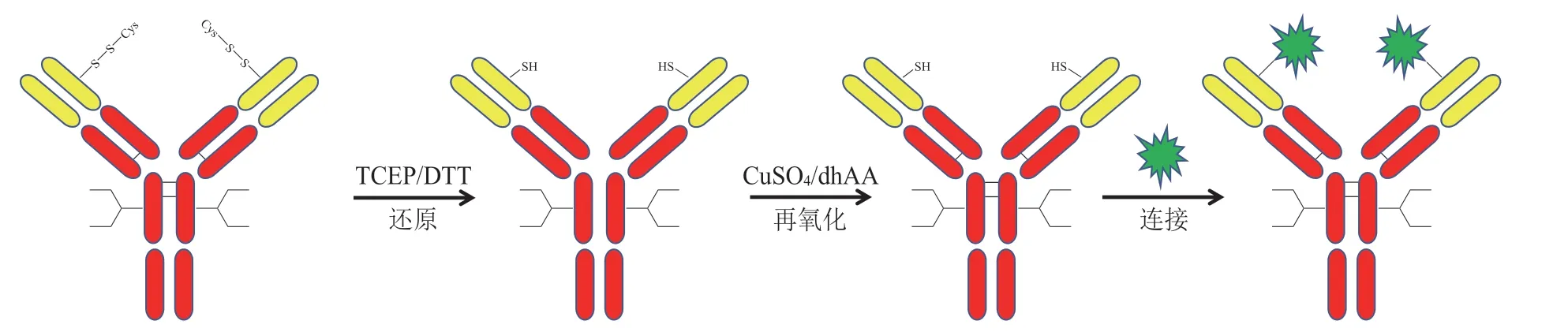
图10 基于Thio-Mab 技术的定点偶联Figure 10 Site-specific conjugation based on Thio-Mab technique
5.3 插入半胱氨酸
基于Thio-Mab 技术的成功,Dimasi 等[28]采用插入半胱氨酸的方法实现抗体和药物分子的定点偶联。在重链第239 位氨基酸后插入半胱氨酸,成功引入和药物连接子反应的巯基,将其与马来酰亚胺基团发生迈克尔加成反应,从而获得ADC(见图11)。该方法引入额外的半胱氨酸可能会导致二硫键错配。
5.4 特殊位点半胱氨酸突变
除上述介绍的利用半胱氨酸进行偶联的方法外,Shiraishi 等[29]和Shinmi 等[30]选择抗体上特殊的位点进行半胱氨酸突变,突变后的抗体有2 个游离巯基。与Thio-Mab 技术相比,该方法省去先还原再氧化的反应流程,可以与含马来酰亚胺基团的药物连接子直接偶联,获得DAR 约等于2 的ADC(见图12)。该方法对抗体序列进行的改造,对突变位点要求较高,需进行大量位点筛选工作。
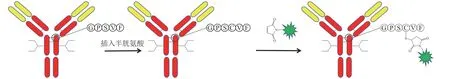
图11 通过插入半胱氨酸实现定点偶联Figure 11 Site-specific conjugation using cysteine insertion
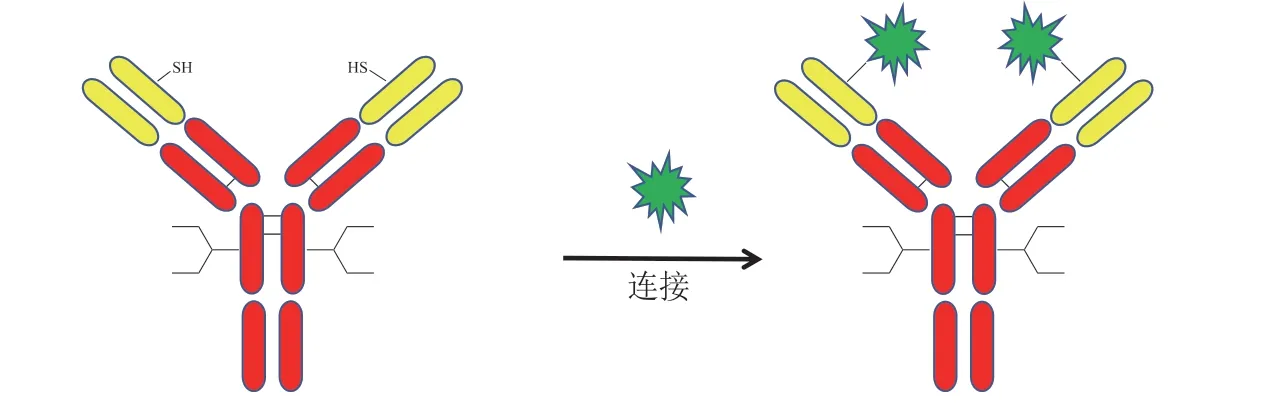
图12 通过特殊位点半胱氨酸突变实现定点偶联Figure 12 Site-specific conjugation using special cystine mutation
6 其他方法
除以上定点偶联方法外,硒代半胱氨酸和丝氨酸代半胱氨酸也可以实现定点偶联。
6.1 硒代半胱氨酸
硒代半胱氨酸与半胱氨酸非常相似,区别是氨基酸结构中的一个硫原子被替换为了硒原子。硒代半胱氨酸的硒醇组比半胱氨酸的硫醇组更亲核,因此抗体可在弱酸性和还原条件下进行偶联,而无需将抗体再氧化。这种在抗体中插入硒代半胱氨酸,获得的工程化抗体被称为“selenomabs”,可以实现在不改变该抗体完整结构的情况下进行区域特异性偶联的目的[31]。
6.2 丝氨酸代半胱氨酸
McDonagh 等[32]开发了丝氨酸代半胱氨酸的方法来实现定点偶联。打开链间二硫键后,利用丝氨酸取代4 或6 个链间半胱氨酸,将反应性半胱氨酸的数量减少到4 或2 个,从而生成均一的ADC。
7 结语
ADC 是肿瘤治疗的研究热点之一,过去10 年间多款ADC 上市,带动ADC 迅速发展,现有多款ADC 处于临床在研阶段。ADC 也开始出现更多形式的单抗,如纳米抗体、抗体Fab 片段、单链可变肽段等;连接子的设计亦在不断改进,越来越多的小分子药物将被用于偶联抗体。随着偶联技术的发展与工艺的完善,ADC 会向着高均一性、高稳定性、高药效的方向发展,癌症治疗的前景必定柳暗花明。
