光度法测定凯氏氮存在的问题研究
2021-04-24周诗雨仇午丹赵晨晨
周诗雨 仇午丹 赵晨晨
(1、浙江人欣检测研究院股份有限公司,浙江 宁波315000 2、浙江中环瑞蓝科技发展有限公司,浙江 宁波315000)
随着生态环境逐渐恶化,湖泊与水体富氧氧化现象普遍,凯氏氮是利用凯氏法对氮含量进行测量,可对水体富营养化程度进行有效评价。运用光度法测定凯氏氮,具有操作简便、快速高效、试剂稳定等优势,从而得到广泛应用。在水样采集完毕后开展实验,在原有技术的基础上加入特定的氢氧化钠,绘制硼酸和氢氧化钠体系校正曲线,使回收率得到极大提升,与实验要求充分满足。
1 凯氏氮测定方法
凯氏氮是利用凯氏法对氮含量进行测量,包括氨氮转化为铵盐后测定的有机氮化合物,该物质包含氨基酸、蛋白质、核算与尿素等,但不含有硝基化合物、叠氮化合物等等。在环境水中蕴含的氮以亚硝酸盐氮、硝酸盐氮为主,当水体受到污染后,导致大量有机氮融入其中,在微生物作用下分解,对水中溶解氧进行消耗,由此导致水质恶化。现阶段,国内饮用水源调查是以凯氏氮为关键指标,如若该项指标超出设计标准,则说明水体受到污染,特别是对水中三种无机氮化合物一同测量时,有利于对水系自净能力、污染程度进行分析,为湖泊与水库富营养化评价提供有利指标[1]。
2 光度法测定凯氏氮实验分析
2.1 仪器与试剂
该实验主要利用型号为U-3410 紫外可见分光光度计、凯氏氮消解装置、蒸馏装置与分析天平。所用试剂包括硫酸钾、氯化铵、硼酸、氢氧化钠、六十酸钾钠,除氯化铵为优级纯度之外,剩余均为分析纯,实验用水为无氨水。
2.2 实验方法
在校正曲线绘制方面,准备8 个容量为50ml 的实验瓶,每个瓶中加入浓度为12%、剂量为10.00ml 的硼酸溶液,与10.00ml 的氢氧化钠溶液,摇晃均匀后,分别按照0、0.5、1.0、2.0、3.0、5.0、7.0、10.0ml 的氯化铵,再将1.0ml 的硫酸钾钠与1.5ml的纳氏试剂加入其中,经过稀释后摇晃均匀,沉淀10 分钟后用比色皿测量吸光度,并绘制标准曲线。在样品分析方面,选择适量的河水样品,将其放入容量为500ml 的凯氏瓶中,将10ml 的浓硫酸、2ml 的硫酸铜、6g 硫酸钾与若干玻璃珠加入其中,摇晃均匀后放在通风柜中加热,直至沸腾,将三氧化硫加入其中使溶液变得清澈,对热源调整后保持微沸状态30min,在常温下冷却后加入250ml 的水,搅拌均匀,当采集蒸馏液200ml 后停止操作[2]。
2.3 结果与讨论
2.3.1 氢氧化钠溶液加入量
采用特定的氯化铵与硼酸溶液量,分别加入不同体积的1mol 氢氧化钠溶液,根据校正曲线实验方式对吸光度进行测定,并对实验现象细致观察,结果如表1 所示。该实验中硼酸用量为推荐值,在50ml 的容量瓶中将浓度为2%、剂量为10ml 的硼酸吸收液加入其中,使其与实际样品条件一致;氯化钠的用量为5.0ml。根据表1 可知,当氢氧化钠溶液量过多或者过少时,胶体溶液均不够稳定,吸光度与溶液加入量之间为正比关系,后者随前者的增加而提升。对此,在实验中应严格把控氢氧化钠的用量。实验表明,当该试剂用量为10ml 时,有色胶体溶液的稳定期最长,可将1mol 的氢氧化钠溶液10ml 与硼酸中和,使胶体处于最佳稳定状态。

表1 NaOH 加入量与吸光度关系
2.3.2 氢氧化钠与硼酸物质量比例与用量
在只改变硼酸与物质量的比例,剩余条件固定的情况下,对胶体稳定性与吸光度的关系进行分析。当二者比例为0.3:1时,吸光度与胶体稳定期呈正比关系。在物质量比例固定不变下,其他溶液加入量变化对吸光度产生更大影响,实验结果如表2。根据该表数据可知,当二者体积发生改变时,对胶体稳定性产生的影响较小,吸光度与二者体积具有正比关系。如若二者体积在0-8ml 之间时,与水样吸收液添加量不符,本次实验在创建校正曲线时,选用浓度为2%,剂量为10ml 的硼酸与10ml 的氢氧化钠溶液作为校正溶液,用于缩短实际样品测定期间的误差。
2.3.3 校正与标准曲线对比
按照本文实验方式绘制校正曲线,与标准曲线一同测定,根据测定结果可知,校正曲线斜率与标准曲线有所区别,因实际样品测定中已经应用了硼酸与氢氧化钠溶液。因此,为了使测定结果更贴近真实值,务必要利用与测定要求充分相符的校正曲线进行分析。在准确性检测中,按照上述方式对某河水样品进行分析,对凯氏氮回收率进行实验。根据结果可知,利用校正曲线测定的凯氏氮含量结果更加可靠,样品回收率在98-100%之间。在精密度检测中,按照上述方式对某河水样品进行分析,开展平行样实验,通过校正曲线法对样品中凯氏氮含量进行测定。
根据实验结果可知,平行实验结果的标准偏差为0.36%,曲线法测定结果精密度更为理想。
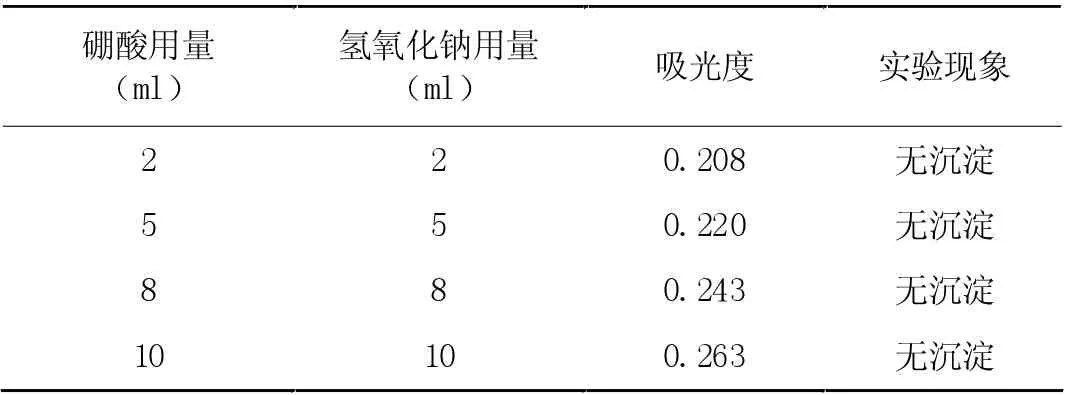
表2 两种溶液用量影响
3 光度法测定凯氏氮问题的修正措施
3.1 取样具有代表性
在天然水源中蕴含多种杂质,根据存在形态与颗粒大小的不同可分为不同形态,即胶体物、悬浮物与溶解物。其中,悬浮物的特点在于动水中以悬浮状态存在,但在静水中根据自重的不同上浮或者下沉。在地面水中无极悬浮物以泥沙为主,还包括矿物质废渣、大粒径粘土等等,此类物质自重较大,容易下沉;有机悬浮物的大小与来源不同,包括水草与小型浮游生物等等,主要源于污水、废水之中;天然水中具有各类蛋白质、粘土胶体等等,前者是有机物分解阶段产生,后者为无机颗粒,可提高水的浑浊度,导致水体变色,如在腐蚀物质影响下使水体变黄或者变为褐色。因此,在实验选样阶段应确保均匀,具有代表性,免受主观因素影响,且样品采集后应及时分析,确保分析结果准确。
3.2 浓缩与消解同步开展
为了提高实验结果准确性,浓缩与消解应同步开展,将操作流程进行简化,由此节约更多时间,只要在上述操作中注重关键环节,针对相同样品实施两次消费与蒸馏,测得值重现性良好。针对相同地点在不同时间内两次取水,采用滴定法对凯氏氮值进行测定,从中选出最佳指标制定最终实验方案。此外,应加强消化阶段的控制。以添加剂用量为主,国际标准为0.2g硫酸铜与6g 无水硫酸钾,二者比例为1:30。如若添加量过多,消化液容易结块,转移难度增加;如若添加量较少,消化时间会延长,还会导致消化不完全。浓硫酸用量应根据样品含水量、重量、蛋白质含量灵活调整。对于大体积、轻重量的产品来说,可适当增加浓硫酸用量,究其原因,此类样品消化中浓硫酸对脱水、消化时的消耗量提升,如若添加浓硫酸量不足,很容易在碳化阶段被烧干。在消化温度方面,可通过循序加热的方式,初始温度为200-300℃,待试样泡沫消失后逐渐提高温度,达到360-600℃。当消化液定容后,在瓶口上加上漏斗,用少量蒸馏水多次洗涤烧瓶与漏斗,减少因误差导致测定结果偏低的情况。在转移完毕后禁止立即定容,而是要将其冷却到室温状态后再定容。
3.3 做好蒸馏工作
实验者应明确预蒸馏的重要意义,在蒸馏器正式应用之前,先将蒸馏水与NaOH 溶液混合起来实施预蒸馏,直到蒸出液无氨为止。仪器每次停用时间超过4 小时便会累积氨气,再次使用时便要进行预蒸馏,确保管内壁附着的氨得到有效冲洗。同时,还应对蒸馏器严密性进行检验。在蒸馏作业中,为避免氨逃逸造成损失。在添加浓NaOH 溶液之前应将冷凝管与蒸馏瓶相连,然后缓慢的将浓碱液注入其中,马上蒸馏。因氨具有较强的挥发性,快速蒸馏可将损失降到最低,使实验结果更加准确可靠。最后,还应对蒸馏温度进行严格控制。蒸馏装置形式较多,大多采用水蒸气蒸馏与直接蒸馏两种形式,根据测定溶液含量与体积的不同,灵活选择常量与半微量蒸馏。当上述两种形式联合应用时,务必要对温度严格控制,如若蒸馏出现暴沸情况,很容易导致吸收液温度提升,导致氨的大量损失,特别是以硼酸为吸收液的情况下,更要注重温度合理。
3.4 加强滴定与测定控制
在实验全过程中滴定属于最后一个环节,其重要性不言而喻,应做好滴定控制工作,否则前面工作将会失去意义。在正式滴定前将溶液瓶摇晃均匀,将酸式滴定瓶冲洗2-3 次后加入盐酸溶液,使内部气泡排除,记录刻度值后正式滴定。在滴定过程中应掌控好速度,接收液从浅绿色朝着无色转变时放慢速度,逐一滴入盐酸标液,到接收液变成浅红色时结束,禁止滴量过多。在空白测定阶段,额外选取两个凯氏瓶,将相同剂量的试样加入其中。根据实践经验可知,空白测定无需每次都开展,可隔一段时间开展一次。但是在更换实际、标准液时必须要开展空白测定。如若空白值超过规定标准,大多是因所用试剂级别不纯、蒸馏水不纯、玻璃器皿清洁不到位所致,需要对原因深入分析。
综上所述,在对水体富营养化评价中,凯氏氮属于重要指标之一。本文利用光度法对凯氏氮进行测定,充分发挥该方法试剂稳定、灵敏性强等特点。根据实验结果表明,氢氧化钠具有增敏效果,并要采用校正曲线保证结果准确性。为了修正实验测定问题,应选取具有代表性的试样,并加强对消化、蒸馏、滴定与测定等环节的控制,才可使实验结果更加准确,更好的解决实际问题。
