谷氨酸钠摄入引起的果蝇肠道菌群多样性变化研究
2021-03-30王丹凤谢甲钰陈文锋
王丹凤,谢甲钰,杨 广*,陈文锋*
(1. 福建农林大学应用生态研究所,福州 350002;2. 福州大学生物科学与工程学院,福州 350108)
谷氨酸钠(Monosodium Glutamate,MSG)是氨基酸的一种钠盐,其是调味料“味精”的主要成分。MSG是一种增味剂,通常添加到食品、蔬菜罐头、汤和加工的肉中。美国食品和药物管理局(FDA)已将MSG归类为“通常被认为是安全的”食品成分,但其使用仍存在争议(Walker and Lupien, 2000)。因此,将MSG添加到食品中时,FDA要求将其列在标签上(Lavine, 2007)。
MSG已被用作食品添加剂数十年。多年来,FDA收到许多有关含MSG食品引起不良反应的传闻。这些反应被称为MSG症候群,包括:头痛、出汗、面部压力或紧绷,脸、颈部和其他区域出现麻木、刺痛或灼痛,快速的、颤动的心跳(心)、胸痛、恶心等(Gehaetal., 2000)。但是,研究人员没有找到有关MSG与这些症状之间联系的明确证据(Jinap and Hajeb, 2010)。不过,研究人员承认,一小部分人可能会对MSG产生短期反应,症状通常较轻,不需要治疗,防止反应的唯一方法是避免食物中含有MSG。
肠道不仅是人体消化吸收的重要场所,同时在维持正常免疫防御功能中发挥着极其重要的作用。作为人体最庞大、最复杂的微生态系统,肠道中存在数量庞大的微生物,肠道菌群及其代谢产物不仅能调节人体健康,而且在膳食和宿主之间起到了重要的桥梁作用(Neish, 2009; Clementeetal., 2012)。但是,关于MSG摄入是否改变肠道菌群组成还未见报道。16S rRNA位于原核细胞核糖体小亚基上,包括10个保守区域(Conserved Regions)和9个高变区域(Hypervariable Regions),其中保守区在细菌间差异不大,高变区具有属或种的特异性,随亲缘关系不同而有一定的差异。因此,16S rDNA可以做为揭示生物物种的特征核酸序列,被认为是最适于细菌系统发育和分类鉴定的指标(Youssefetal., 2009; Caporasoetal., 2011; Hessetal., 2011)。16S rDNA扩增子测序(16S rDNA Amplicon Sequencing),通常是选择某个或某几个变异区域,利用保守区设计通用引物进行PCR扩增,然后对高变区进行测序分析和菌种鉴定。16S rDNA扩增子测序技术已成为研究环境样品中微生物群落组成结构的重要手段(Youssefetal., 2009; Caporasoetal., 2011; Hessetal., 2011)。黑腹果蝇作为重要的模式生物,与高等动物相比,存在很高比例的同源基因,一直是各种生物学问题研究很好的模式(Uguretal., 2016; Staatsetal., 2018)。本文利用黑腹果蝇作为材料,利用16S rDNA测序研究其肠道响应MSG摄入的微生物组成变化,以期为理解MSG摄入对肠道菌群的影响提供更多的依据。
1 材料与方法
1.1 果蝇培养及处理
本研究所用果蝇为行为实验常用的果蝇,基因型为w1118(Bloomington果蝇保种中心编号为BS5905)。饲养于含有0.75%大豆粉、4.5%玉米粉、1.5%酵母、0.5%丙酸、0.1%尼泊金甲酯、0.02%玉米糖浆、1.25%蔗糖、1.25%葡萄糖、0.5%琼脂的食物中。饲养条件为25℃,光周期L ∶D=12 h ∶12 h。收集羽化后2~3 d的果蝇30头,用含有或不含有1% MSG(上海生物工程生物有限公司,A602012)的蔗糖培养基(5%蔗糖,1%琼脂进行饲养(培养基经过高温高压灭菌处理),第4天的Zeitgeber Time 2(ZT2)(ZT0为开灯时间,ZT12为关灯时间)收集果蝇腹部样品并-80℃冻存。所有化学试剂采购自国药集团化学试剂有限公司或上海生工生物工程股份有限公司。
1.2 测序样品准备及测序分析
采用通用型柱式基因组DNA提取试剂盒对基因组DNA进行提取(北京康为世纪生物科技有限公司,CW2298),按照说明书操作从果蝇腹部提取总DNA。提取裂解液中加入终浓度为0.1 mg/mL的溶菌酶(北京康为世纪生物科技有限公司,CW0887S)。利用琼脂糖凝胶电泳检测DNA的纯度和浓度,取适量的样本DNA于离心管中,使用无菌水稀释样本至1 ng/μL。以稀释后的基因组DNA为模板,使用带标签序列(Barcode)的16S V4区特异引物(515F:5′-GTGCCAGCMGCCGCGG TAA-3′和806R:5′-GGACTACHVHHHTWTCTAAT-3′)(Caporasoetal., 2011),Phusion©High-Fidelity PCR Master Mix with GC Buffer和高效高保真酶进行PCR(ThermoFisher, F532S),确保扩增效率和准确性。PCR产物使用2%琼脂糖凝胶进行电泳检测;根据PCR产物浓度进行等量混样,充分混匀后使用1×TAE 2%琼脂糖胶电泳纯化PCR产物,剪切回收目标条带。使用GeneJET胶回收试剂盒回收产物(Thermo Fisher, K0629)。最后使用Ion Plus Fragment Library Kit 48 rxns建库试剂盒(ThermoFisher, 4471252)进行文库的构建,构建好的文库经过Qubit(ThermoFisher, Q33266)定量和文库检测合格后,使用的Ion S5TMXL(ThermoFisher, A27214)进行上机测序。
1.3 测序数据的处理
使用Cutadapt(V1.9.1, http://cutadapt.read thedocs.io/en/stable/)(Asshaueretal., 2015)先对测序读值(Reads)进行低质量部分剪切,再根据Barcode从得到的Reads中拆分出各样品数据,截去Barcode和引物序列初步质控得到原始数据(Raw Reads)。经过以上处理后得到的Reads进行嵌合体序列去除,Reads序列通过(https://github.com/torognes/vsearch/)与物种注释数据库进行比对检测嵌合体序列,并去除其中的嵌合体序列(Rognesetal., 2016),得到最终的有效数据(Clean Reads)。
1.4 OTU聚类和物种注释
高通量测序得到的16S序列有成千上万条,如果对每条序列都进行物种注释的话,工作量大、耗时长,而且16S扩增、测序等过程中出现的错误会降低结果的准确性。因此,在16S分析中引入分类操作单位(Operational Taxonomic Units, OTUs),对相似性序列进行聚类,分成数量较少的分类单元,基于分类单元进行物种注释。这不仅简化工作量,提高分析效率,而且OTU在聚类过程中会去除一些测序错误的序列,提高分析的准确性。利用Uparse软件(Uparse v7.0.1001,http://www. drive5.com/uparse/)(Haasetal., 2011)对所有样品的全部Clean Reads进行聚类,默认以97%的一致性(Identity)将序列聚类成为OTUs,同时选取OTUs的代表性序列,依据其算法原则,筛选OTUs中出现频数最高的序列作为OTUs的代表序列。对OTUs序列进行物种注释,用Mothur方法与SILVA132(http://www.arb-silva.de/)(Edgar, 2013)的SSUrRNA数据库(Wangetal., 2007)进行物种注释分析(设定阈值为0.8~1),获得分类学信息并分别在各个分类水平:界(Kingdom),门(Phylum),纲(Class),目(Order),科(Family),属(Genus),种(Species)统计各样本的群落组成。使用MUSCLE(Quastetal., 2013)(Version 3.8.31, http://www.drive5. com/muscle/)软件进行快速多序列比对,得到所有OTUs序列的系统发生关系。最后对各样品的数据进行均一化处理,以样品中数据量最少的为标准进行均一化处理,后续的α多样性分析和β多样性分析都是基于均一化处理后的数据。
1.5 样本复杂度分析(α多样性)
α多样性(αDiversity)用于分析样本内(Within-community)的微生物群落多样性(Lietal., 2013),通过单样本的α多样性分析可以反映样本内的微生物群落的丰富度和多样性,包括用一系列统计学分析指数、物种多样性曲线等来评估各样本中微生物群落的物种丰富度和多样性的差异。稀释曲线(Rarefaction Curve)和等级聚类曲线(Rank Abundance)是常见的描述组内样本多样性的曲线。稀释曲线是从样本中随机抽取一定测序量的数据,统计它们所代表物种数目(即OTUs数目),以抽取的测序数据量与对应的物种数来构建曲线。稀释曲线直接反映测序数据量的合理性,并间接反映样本中物种的丰富程度,当曲线趋向平坦时,说明测序数据量渐进合理,更多的数据量只会产生少量新的物种(OTUs)。等级聚类曲线是将样本中的OTUs按相对丰度(或者包含的序列数目)由大到小排序得到对应的排序编号,再以OTUs的排序编号为横坐标,OTUs中的相对丰度(也可用该等级OTU中序列数的相对百分含量)为纵坐标,将这些点用折线连接,即绘制得到Rank Abundance曲线,它可直观的反映样本中物种的丰富度和均匀度。在水平方向上,物种的丰富度由曲线的宽度来反映,物种的丰富度越高,曲线在横轴上的跨度越大;在垂直方向上,曲线的平滑程度,反映了样本中物种的均匀程度,曲线越平缓,物种分布越均匀(Lundbergetal., 2013)。使用Qiime软件(Version 1.9.1)计算Observed-otus、Chao1、Shannon、Simpson、ACE、Goods-coverage和PD_whole_tree指数,使用R软件(Version 2.15.3)绘制稀释曲线和Rank abundance曲线,并使用R软件进行α多样性指数组间差异分析;α多样性指数组间差异分析进行非参数wilcox检验。
1.6 多样本比较分析(β多样性)
β多样性(βDiversity)是对不同样本的微生物群落构成进行比较分析。首先根据所有样本的物种注释结果和OTUs的丰度信息,将相同分类的OTUs信息合并处理得到物种丰度信息表(Profiling Table)。同时利用OTUs之间的系统发生关系,进一步计算Unifrac距离(Unweighted Unifrac)(Lozuponeetal., 2007; Lozuponeetal., 2011)。Unifrac距离是一种利用各样本中微生物序列间的进化信息计算样本间距离,两个以上的样本,则得到一个距离矩阵。然后,利用OTUs的丰度信息对Unifrac距离(Unweighted Unifrac)进一步构建Weighted Unifrac距离(Lozuponeetal., 2007)。最后,通过多变量统计学方法主成分分析(PCA, Principal Component Analysis)、主坐标分析(PCoA, Principal Co-ordinates Analysis)、无度量多维标定法(NMDS, Non-Metric Multi-Dimensional Scaling)等方法,从中发现不同样本(组)间的差异。用Qiime软件(Version 1.9.1)计算Unifrac距离。使用R软件(Version 2.15.3)绘制主成分分析(PCA, Principal Component Analysis),主坐标分析(Principal Co-ordinates Analysis, PCoA)和无度量多维标定法(Non-Metric Multi-Dimensional Scaling, NMDS)图。PCA分析使用R软件的ade4包和ggplot2软件包,PCoA分析使用R软件的WGCNA、stats和ggplot2软件包,NMDS分析使用R软件的vegan软件包。
2结果与分析
2.1 测序数据的质量
本研究选择16S V4区对对照组(Control)和MSG饲喂组(MSG)果蝇肠道菌群进行扩增测序,将IonS5TMXL下机数据导出fastq文件。根据barcode序列区分各个样本数据。然后进行嵌合体过滤,得到可用于后续分析的有效数据,即Clean Reads。数据处理过程中各步骤得到的序列统计结果见表1。可以看出,所有样品中Clean Reads中碱基质量值大于20(测序错误率小于1%)的碱基所占的百分比均大于80%,Clean Reads的数目与Raw Reads数目的百分比均在90%以上,说明测序结果准确,整体数据质量比较高(表1)。
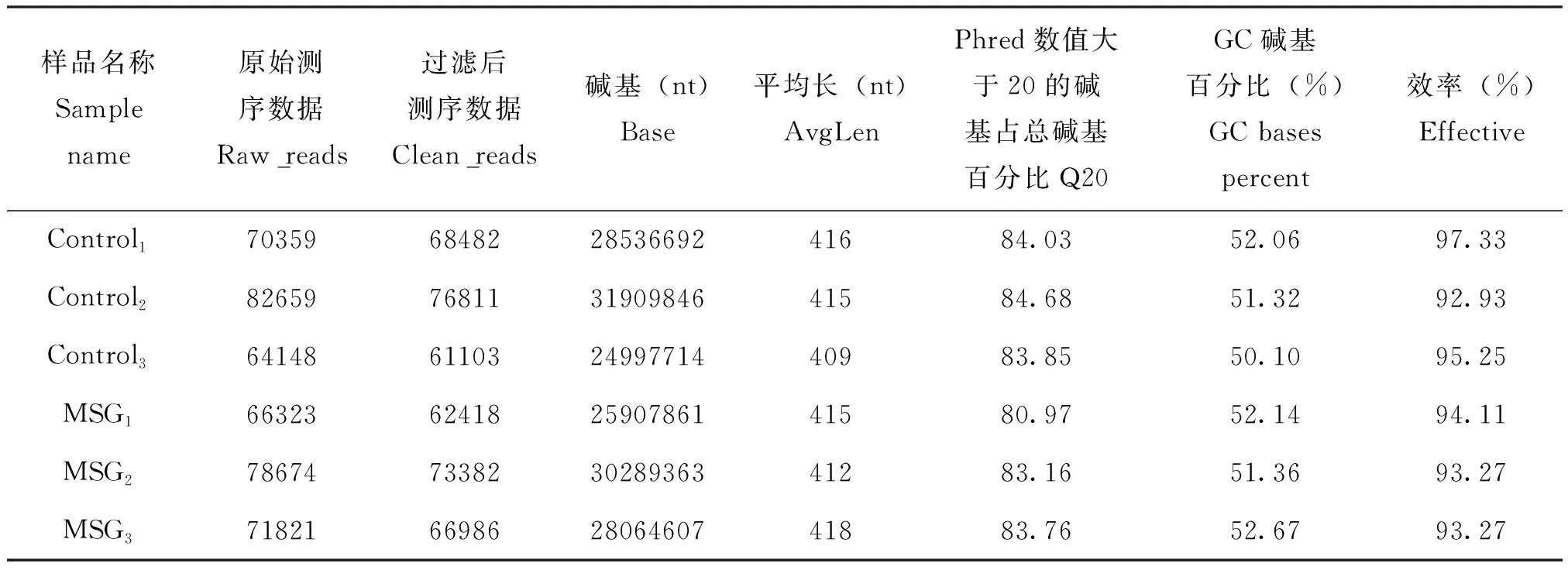
表1 测序数据预处理统计及质控
2.2 MSG摄入对肠道菌群组成的影响
为研究各样本的肠道菌群物种组成,对所有样本的有效序列(Effective Tags),以97%的一致性(Identity)进行OTUs聚类,然后对OTUs的代表序列进行物种注释。为了快速直观的展示样本中的物种组成及丰度信息,本研究构建了OTUs对应的物种注释热图,以每个OTUs大于1 000个读值进行过滤,发现MSG组肠道菌群更为复杂(图1A)。同时,本研究使用KRONA(Ondovetal., 2011)对物种注释结果进行可视化展示,也得到类似结果(图1B-C)。这些结果显示:与对照组相比,MSG饲喂组肠道菌群组成确实发生了明显变化,MSG组肠道菌群组成更为复杂(图1)。
2.3 MSG对肠道菌群物种分布的影响
根据物种注释结果,本研究选取每个样本在各分类水平(门Phylum、纲Class、目Order、科Family、属Genus、种Species)上最大丰度排名前10的物种,生成物种相对丰度柱形累加图,以便直观查看各样本在不同分类水平上,相对丰度较高的物种及其比例。结果显示MSG饲喂组相比对照组,厚壁杆菌门Firmicutes、芽孢杆菌纲Bacilli、乳杆菌目Lactobacillales、乳杆菌科Lactobacillaceae丰度显著升高,而属种水平差异从该分析上看不出,被归为其它类(图2)。

图2 各分类水平上的物种相对丰度柱形图Fig.2 Histogram of species relative abundance at each classification level注:横坐标是样本名;纵坐标表示相对丰度;Others表示图中这10个门之外的其他所有门的相对丰度之和。Note: The abscissa was the sample name; the ordinate represented the relative abundance. Others represented the sum of the relative abundances of all other groups except 10 groups in the figure.

图3 物种丰度聚类图Fig.3 Cluster map of species abundance注:纵向为样本信息,横向为物种注释信息,图中左侧的聚类树为物种聚类树;热图对应的值为每一行物种相对丰度经过标准化处理后得到的Z值,即一个样本在某个分类上的Z值为样本在该分类上的相对丰度和所有样本在该分类的平均相对丰度的差除以所有样本在该分类上的标准差所得到的值。Note: The sample information was in the vertical direction, and the species annotation information was in the horizontal direction. The cluster tree on the left in the figure was the species cluster tree. The corresponding value of the heat map was the Z value obtained by normalizing the relative abundance of the species in each row. The Z value of a category was the difference between the relative abundance of the samples in the category and the average relative abundance of all samples in the category divided by the standard deviation of all the samples in the category.
于是,根据对照组和MSG饲喂组不同分组在各分类水平(门Phylum、纲Class、目Order、科Family、属Genus)上的物种注释及丰度信息,本研究进一步选取丰度排名处于前35的信息,根据其在每个分组中的丰度信息,从物种和分组两个层面进行聚类,绘制成热图,便于发现哪些物种在哪个分组中聚集较多或含量较低。结果也显示MSG饲喂组相比对照组,厚壁杆菌门Firmicutes、芽孢杆菌纲Bacilli、乳杆菌目Lactobacillales、乳杆菌科Lactobacillaceae、乳杆菌属Lactobacillus丰度显著升高(图3)。
另外,本研究还利用LEfSe(LDA Effect Size)分析群落结构差异,找出MSG饲喂组和对照组间丰度变化差异显著的物种及其在两个分组间的富集情况。LEfSe的统计结果包括三部分,分别是LDA值分布柱状图,进化分支图(系统发育分布)和组间具有统计学差异的Biomarker在不同组中丰度比较图(Segataetal., 2011)。LDA值分布柱状图显示MSG组和对照组间具有统计学差异的Biomarker共12个,其中影响最大的差异物种为乳杆菌目Lactobacillales和变形菌门Proteobacteria(图4 A)。进化分支图显示两个重要分支为厚壁杆菌门Firmicutes和变形菌门Proteobacteria,厚壁杆菌门中的重要成员就包括芽孢杆菌纲(Bacilli)、乳杆菌目Lactobacillales和肠杆菌科Enterococcaceae(图4 B)。与前面结果一致,LEfSe丰度比较图也显示MSG饲喂组相比对照组,厚壁杆菌门、芽孢杆菌纲Bacilli、乳杆菌目丰度显著升高(图4 C)。

图4 物种丰度的LEfSe分析Fig.4 LEfSe analysis of species abundance注:LDA值分布柱状图中展示了LDA Score大于设定值(默认设置为4)的物种,即组间具有统计学差异的Biomarker。展示了不同组中丰度差异显著的物种,柱状图的长度代表差异物种的影响大小(即为LDA Score)。在进化分支图中,由内至外辐射的圆圈代表了由门至属(或种)的分类级别。在不同分类级别上的每一个小圆圈代表该水平下的一个分类,小圆圈直径大小与相对丰度大小呈正比。着色原则:无显著差异的物种统一着色为黄色,差异物种Biomarker跟随组进行着色,红色节点表示在红色组别中起到重要作用的微生物类群,绿色节点表示在绿色组别中起到重要作用的微生物类群,若图中某一组缺失,则表明此组中并无差异显著的物种,故此组缺失。图中英文字母表示的物种名称在右侧图例中进行展示。Note: The LDA value distribution histogram showed the species with an LDA Score greater than the set value (the default setting was 4), that was, biomarker with statistical differences between groups. Species with significantly different abundances in different groups were shown, and the length of the histogram represented the magnitude of the impact of the different species (ie, LDA Score). In the evolutionary branch diagram, the circle radiating from the inside to the outside represented the classification level from the door to the genus (or species). Each small circle at a different classification level represented a classification at that level, and the diameter of the small circle was proportional to the relative abundance. Coloring principle: The species with no significant difference were uniformly colored yellow, and the different species Biomarker followed the group for coloring. The red node indicated the microbial group that played an important role in the red group, and the green node indicated that it played an important role in the green group. Microbial groups, if a group was missing in the picture, it means that there were no significant differences in species in this group, so this group was missing. The names of the species indicated by the English letters in the figure were shown in the legend on the right.
2.4 MSG对肠道菌群α多样性的影响
本研究首先对不同样本在97%一致性阈值下的α多样性分析指数(Shannon、Simpson、Chao1、ACE、Goods_coverage、PD_whole_tree)进行统计,Shannon和Simpson指数统计结果显示MSG饲喂组和对照组相比,微生物群落多样性组成存在显著差异(表2)。另外,本研究还通过稀释曲线和等级聚类曲线来分析物种丰富度和多样性的差异。与α多样性分析指数结果一致,物种多样性曲线分析也显示MSG饲喂组和对照组相比,微生物群落多样性组成存在差异(图5)。
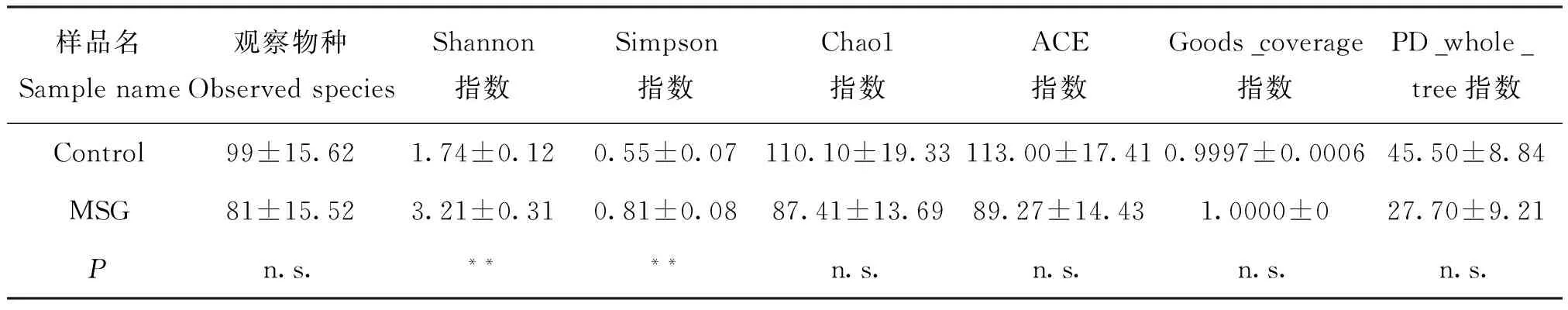
表2 α多样性指数统计表

图5 稀释曲线和等级聚类曲线Fig.5 Rarefaction and rank curves
2.5 MSG对肠道菌群β多样性的影响
基于Weighted Unifrac或Unweighted Unifrac距离的PCoA分析显示MSG饲喂组和对照组微生物群落存在显著差异(图6)。基于OTUs水平的主成分PCA分析和无度量多维标定NMDS分析也得到类似结果(图6)。

图6 β多样性分析Fig.6 β diversity analysis
3 结论与讨论
MSG摄入可显著引起果蝇肠道菌群组成变化。长期饮食摄入会影响宿主肠道中微生物的结构和活性,且可快速、可重复地改变宿主肠道菌群(Davidetal., 2014)。反之,肠道菌群也可快速反馈与饮食结构相关的变化,促进宿主饮食方式的多样性,影响宿主营养状况。例如,果蝇肠道细菌摄入的糖决定了果蝇的脂质含量,与无菌果蝇相比,肠道含有醋杆菌的果蝇脂质含量更低(Huang and Douglas, 2015)。本研究首次证明MSG的摄入可以显著引起果蝇肠道菌群组成变化,为理解MSG摄入对生物个体的影响提供了新的研究思路。
MSG摄入显著提高了果蝇肠道乳杆菌属Lactobacillus的组成。果蝇的肠道菌群至少包含 5~20种细菌,其中乳杆菌属Lactobacillus和醋杆菌属Acetobacter是实验室饲养的果蝇肠道菌群的优势菌群(Wongetal., 2013)。乳杆菌属Lactobacillus也是最常见的人类益生菌(Walter, 2008)。肠道是暴露于影响宿主生理的环境信号的主要管道,并通过神经元和血淋巴连接到大脑,肠道菌群会影响包括大脑在内的许多器官的功能。诸多研究表明,肠道与大脑之间的双向交流可以影响包括焦虑、认知、伤害感受和社交互动在内的多种行为(Diaz Heijtzetal., 2011)。最近一项研究证明乳杆菌在果蝇中可以通过细菌木糖异构酶调节果蝇运动行为,并确定了章鱼胺能神经元在其中起到媒介作用(Schretteretal., 2018),揭示了肠道菌群在调节运动中的新作用。本研究揭示MSG摄入显著提高了果蝇肠道乳杆菌属Lactobacillus的组成,其功能有待进一步研究。
不同物质摄入对肠道菌群影响各异。比如高盐摄入可提高老鼠肠道克里斯滕森菌科Christensenellaceae和棒状杆菌科Corynebacteriaceae的含量(Bieretal., 2018)。低脂摄入提高人类肠道中布劳特氏菌Blautia和栖粪杆菌属Faecalibacterium水平,而高脂摄入增加另枝菌属Alistipes和拟杆菌属Bacteroides水平,降低栖粪杆菌属Faecalibacterium水平(Wanetal., 2019)。MSG对肠道乳杆菌的影响有待在其它物种中进一步探讨。
