The active ingredients of Huanglian Jiedu Decoction in treating COVlD-19 based on network pharmacology,molecular docking and molecular dynamics simulation
2021-02-04PingXieXiaohanJinYuanfaLaiChunliHaoHuabinHuangShaoguiHeQihuaYou
Ping Xie,Xiaohan Jin,Yuanfa Lai*,Chunli Hao,Huabin Huang,Shaogui He,Qihua You
1 College of Environment and Public Health,Xiamen Huaxia University,Xiamen 361024,Fujian,China.
2 Biochemical Pharmacy Engineering Research Center of Fujian Province University,Xiamen 361024,Fujian,China.
3 Xiamen Key Laboratory of food and drug safety,Xiamen 361024,Fujian,China.
Abstract Objective: To explore the active ingredients of Huanglian Jiedu Decoction (HLJD) for the treatment of COVID-19 and to further verify the combination mode.Methods: The TCMSP database was used to search for HLJD active ingredients and targets.COVID-19 targets were collected from GeneCards,DisGeNET and OMIM databases.Material-active-ingredients-targets (gene)network and targets protein-protein interaction network were constructed using Cytoscape 3.8.0 and the STRING database.GO functional enrichment analysis and KEGG pathway enrichment analysis of core targets were performed using R software.Cytoscape 3.8.0 was used to build “compound-targets-pathways” to predict HLJD mechanisms,and active ingredients were used as ligands to molecularly dock with SARS-CoV-2 3CL hydrolase,Spike glycoprotein and ACE2.The binding energy was calculated by molecular dynamics simulations and molecular mechanics Poisson-Boltzmann surface area method,and intermolecular interactions and the contribution of each residue to the binding free energy were analyzed.Results: Four medicinal materials,66 compounds and 219 targets were identified.It is found that the Protein-Protein Interaction core network contained 35 HLJD key targets proteins for COVID-19 treatment.705 GO functional enrichment entries (P < 0.05) were produced; while KEGG pathway enrichment analysis identified 142 pathways (P < 0.05) involving the Tumor Necrosis Factor signaling pathway and Interleukin-17 signaling pathway,etc.The binding energies of Kihadanin A,Palmidin A,Obacunone and Hispidone are much smaller than those of the currently reported clinical drugs with anti-SARS-CoV-2 drugs.The results of the binding energy indicate that van der Waals force is the main driving force for enzyme-substrate combination,whereas the electrostatic interaction and non-polar solvents contribute less.Conclusion: The “multi-component-multi-targets-multi-pathway” synergy of HLJD,which binds to SARSCoV-2 3CL hydrolase,Spike glycoprotein and ACE2,can act on targets Heat Shock Protein 90 Alpha Family Class A Member 1,Adrenoceptor Beta 2,Checkpoint Kinase 1,Peroxisome Proliferator-Activated Receptor Gamma and Mitogen-activated protein kinase 14 to regulate multiple signal pathways,and it may have a therapeutic effect on COVID-19.
Keywords: Network pharmacology,Molecular docking,Molecular dynamics simulation,Huanglian Jiedu Decoction,COVID-19
Background
Since December 2019,the raging coronavirus disease 2019 (COVID-19) arising out of the SARS-COV-2 across China,the United States,Brazil,Russia,and many other countries has been posing a serious threat to global public health [1-2].COVID-19 is mainly spread through face-to-face inhalation and the exhalation of respiratory droplets; thus,everyone is susceptible to it.Besides,it can be transmitted when people touch contaminated surfaces.Due to such rapid spreading,COVID-19 has been listed as a highly contagious infectious disease.At present,the outbreak of COVID-19 in China has been contained,but the situation globally is deteriorating,and the number of confirmed cases is still on the rise.Based on reported clinical studies,patients with mild symptoms may develop fever,coughing,polypnea,and dyspnea.In addition to cardiac trauma,patients with severe symptoms often suffer cerebral injury,hepatic injury,impaired renal function and respiratory failure,or even death [3].Despite the absence of effective drugs against SARS-CoV-2,traditional Chinese medicine (TCM) has played a critical role in resisting plagues since ancient times,which owes to its multi-components,multitargets and less side effect.Therefore,TCM has been widely used in the fight against COVID-19.
Diagnosis and Treatment Protocol for Novel Coronavirus Pneumonia (Trial Version 7) [4] issued by the Chinese government explicitly stated that people catch novel coronavirus pneumonia (NCP) because they have been exposed to the virus that acts on the lungs.To be specific,the mild types consist of: (1)pulmonary stagnation from noxious dampness; (2)pulmonary obstruction from cold dampness; the severe types are: (1) pulmonary obstruction from epidemic toxin; (2) overabundant heat at both Qifen and Yingfen,which are part of a syndrome differentiation method used to signify the extent and damage of a febrile disease; the critical type is only identified with the syndrome of the internal blockade and external collapse.HLJD was originally reported in The Handbook of Prescriptions for Emergencies written by Hong GE [5],but its name first appeared in Waitai Miyao (Arcane Essentials from the Imperial Library).HLJD has the functions of purging fire,detoxification,and releasing three-cavity fire toxins in which four drugs complement each other,making it one of the classical prescriptions for fire purging and detoxification.In particular,Diagnosis and Treatment Protocol for Novel Coronavirus Pneumonia (Trial Version 3) [6] has recommended several compound TCM prescriptions for the diagnosis and treatment of COVID-19,including HLJD.According to the pooled analysis of registered clinical trial protocols of TCM for the prevention and treatment of COVID-19 authored by YU [7],HLJD was explicitly listed in TCM treatment protocols against COVID-19 issued by the Health Commission and Traditional Chinese Medicine Administration of Hebei Province,Jilin Provincial Pneumonia Expert Panel against COVID-19 with TCM,Sichuan Provincial Administration of TCM,Guangxi Zhuang Autonomous Region Administration of TCM,Health Commission of Yunnan Province and Health Commission of Hunan province; Besides,as mentioned in Registered Clinical Trial Protocols of TCM for the Prevention and Treatment of COVID-19 released by the team of professor Boli ZHANG and professor Qingquan LIU [8] on February 6,2020,HLJD was extensively used in the treatment of COVID-19.
Network pharmacology reveals the interconnections between “components-targets-pathway-disease” at a system level.Due to its similarities with TCM in terms of multi-component,multi-target,and multi-pathway characteristics,network pharmacology is also widely explored for researching compound TCM prescriptions;Focusing on the interactions between a large number of drug molecules and receptor proteins,molecular docking is a computational approach that attempts for the prediction of receptor-ligand binding modes and binding affinities as well [9].Research has shown that[10] SARS-CoV-2 enters into the host cell through the interaction of its Spike glycoprotein with the host receptor ACE2.Furthermore,SARS-CoV-2 3CL hydrolase is the core component of the hydrolase precursor in the process of virus replication,which is considered to be an effective therapeutic target.In the light of molecular docking results,molecular dynamics simulations are performed for the best protein molecule complex,and then the binding free energy is calculated by the molecular mechanics Poisson-Boltzmann surface area (MM-PBSA) method to analyze the driving force for the binding of compounds,thus revealing the amino acids that play a key role in the process of such binding.In this review,the active compounds of HLJD for the treatment of COVID-19 are explored by means of integrated multiple computational methods,such as network pharmacology,molecular docking,and molecular dynamics simulation.These results provide a reference for clinical diagnosis and treatment of COVID-19 as well as for an in-depth study of HLJD.
Materials and methods
Screening of active compounds and Relevant of HLJD
With the help of the Traditional Chinese Medicine Systems Pharmacology (TCMSP; http://tcmspw.com/)[11],the main components and targets of the candidate HLJD were obtained,such asCoptidis Rhizoma(CR),Scutellaria Root(SR),Cortex Phellodendri(CP) andFructus Gardeniae(FG).In the meantime,since druglikeness (DL) and oral bioavailability (OB) are very important pharmacokinetic parameters for HLJD as an oral drug,the candidate compounds with OB ≥ 30%and DL ≥ 0.18 were identified as active compounds[12].Next,the Universal Protein Resource (UniProt;https://www.uniprot.https.org/) was utilized to convert the protein names of the targets into their gene names.
Establishment of “material-active-ingredientstargets” network
The materials-active-ingredients-targets (gene)network and targets’ protein-protein interaction (PPI)network were constructed by using Cytoscape 3.8.0,and relevant analyses were performed.
Searching related targets of COVID-19 and Construction of protein-protein interaction (PPI)network
By searching the keywords like “novel-coronaviruspneumonia” in the databases including GeneCards(https://www.genecards.org/),DisGeNE(https://www.disgenet.org/) and OMIM(https://omim.org/),the disease targets of COVID-19 were acquired; by removing the repetitive targets and using the UniProt database for correction and integration,common genes for the active compounds of HLJD and the disease targets of COVID-19 were obtained through the construction of the Venn 2.1.0 diagram.Furthermore,these common targets were imported into the STRING database [13].By restricting the species to human beings,and taking into account the proteins with high confidence (0.9) in this study,isolated targets were removed to construct the PPI network and conduct relevant analyses.
Targets enrichment analysis
All analyses were performed with the R software,and the threshold for statistical significance was set atP<0.05.Furthermore,GO enrichment and KEGG pathway enrichment were employed for the analysis of core targets.After that,the bar and bubble charts were mapped through R software to further analyze the biological process and key signaling pathways of HLJD against COVID-19.
Construction of ingredients-targets-common pathways network
Based on the top 20 critical key signaling pathways of HLJD for treatment of COVID-19,the networks of“compounds-targets” and “targets-pathways” were constructed.These two networks were then merged into the “compounds-targets-common pathways” network with the help of Cytoscape 3.8.0 in which a higher Degree value means that it plays a greater role.
Molecular docking and virtual screening
The potential active compounds were downloaded from the TCMSP database and stored in mol2 format.Then,supported by chem3D,energy forms were minimized and converted to PDB format.Further,by importing these active compounds into AutoDock Tools (v.1.5.6) and adding hydrogen,all rotatable bonds in the molecule were set for flexible docking,then were saved as PDBQT format.Next,threedimensional (3D) structures of the SARS-CoV-2 3CL hydrolase (PDB ID: 6LU7),Spike glycoprotein (PDB ID: 6VSB) and ACE2 (PDB ID: 1R42) in the PDB form were downloaded from a PDB database(https://www.rcsb.org/).By removing water molecules and original ligand,and adding hydrogen atoms,these structures were subsequently saved in pdbqt format.Then,using the potential active compounds data sets as ligands,and the target proteins as receptors,in combination with their action sites,the coordinates and length,width,height of the Grid Box were determined.Finally,by means of AutoDock Vina 1.1.2 and Python script,batches of molecular docking [14] were carried out,and the docking results were analyzed.
Molecular dynamics simulation of the complexes
The best binding conformations of the Kihadanin A and the SARS-CoV-2 3CL hydrolase complexes from the molecule docking program were selected and prepared for molecular dynamics simulation with Gromacs 2019.6 [15].The system was solvated with spc216 water molecules.In order to neutralize the system,four sodium ions were added to the box.In this experiment,the Amber99sb-ildn force field was used for SARSCoV-2 3CL hydrolase,while GAFF force field was adopted for the Kihadanin A.By maintaining the temperature of the system at 300K,the selected complexes of protein and small molecular ligand were first placed into the center of a cuboid box with periodic boundary conditions.The minimum distance between any atom of the protein and the box wall was maintained at 1.0 nm.After that,the energy minimization was executed by using the steepest descent method for 1,000 steps.Then,the optimized system equilibration was performed gradually for 100 ps at the constant number of particles,Volume and Temperature (NVT) ensemble,and then switched to the constant Number of Particles,Pressure and Temperature (NPT) ensemble for equilibrating another 100 ps.During this process,the position of the systems was restricted as well.Finally,a 50 ns molecular dynamics simulation was performed with the whole system at a constant temperature of 300 K.
Calculation of the binding free energy by MMPBSA
The binding free energy of receptor-ligand complexes after molecular dynamics simulation was calculated by MM-PBSA [16-17].Binding free energy of the Kihadanin A and the SARS-CoV-2 3CL hydrolase complexes was calculated by using the G_mmpbsa tool.The lower binding free energy indicated the higher affinity between receptor and ligand [18].By decomposing the total binding free energy of the system into various residues,the contributions of various interactions could be obtained.
Results
Screening of active compounds
After searching through the TCMSP database,a total of 429 main compounds were obtained,including 48 ones from CR,143 ones from SR,140 ones from CP and 98 ones from FG.On this basis,85 active compounds were screened out under the conditions of OB ≥ 30% and DL ≥ 0.18,including 14 from CR,36 from SR,37from CP,and 15 from FG.
“Material-compounds-targets” network
The “materials-compounds-targets” network consisted of 285 nodes and 1,427 edges.Key active compounds and targets were found with more connections,so they played more significant roles as the hubs.In this network,it was found that,each active compound was connected to 14.54 targets,and each target was linked to 4.38 active compounds on an average basis,revealing the synergistic effects of multiple components and multiple targets.According to the degree analysis,the top five compounds were MOL000098-quercetin,MOL000422-kaempferol,MOL000173-wogonin,MOL002714-baicalein and MOL000790-isocorypalminei,which interacted with 140,55,42,34,and 31 target proteins,respectively.With regard to the targets,the top five in degree were Prostaglandin-Endoperoxide Synthase 2 (PTGS2),Prostaglandin-Endoperoxide Synthase 2 (PTGS1),HSP90AA1,Nuclear Receptor Coactivator 2 (NCOA2),and Sodium Voltage-Gated Channel Alpha Subunit 5(SCN5A),which interacted with 54,43,42,32,and 32 compounds,respectively.The compounds in CP,CR,SR and FG were represented in light purple,lake blue,blue and yellow,respectively,while targets were indicated in pink,as shown in Figure 1.
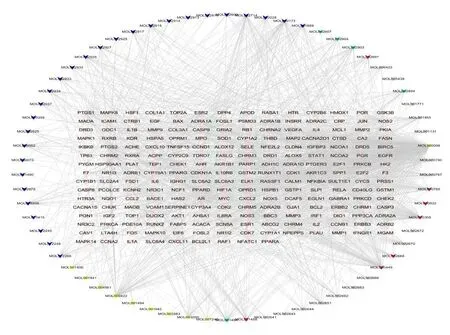
Figure 1 “Material-active ingredients-targets (gene)” network diagram of HLJD

Figure 2 Venn diagram of the therapeutic targets of HLJD

Figure 3 The protein-protein interaction network
Common targets and Protein-Protein Interaction(PPI) Network Construction
A total of 254 COVID-19 related disease targets were obtained after removing the repeated ones.Then,these 254 disease targets and 205 therapeutic targets of HLJD were put into Venny 2.1.0 for interactions,and the results were shown in Figure.2.The PPI network diagram was analyzed by utilizing the STRING platform (https://string-db.org/cgi/input.pl),and the network data were downloaded and imported into Cytoscape.A total of 35 key target proteins for the HLJD treatment of COVID-19 were finally obtained from the analysis of the network topology parameters.Among them,RELA Proto-Oncogene (RELA),MAPK3,MAPK1,Interleukin-6 (IL-6) and MAPK14 had a decreasing degree of freedom,as shown in Figure 3.
GO enrichment analysis and KEGG pathway analysis
GO enrichment analysis was conducted on potential targets.A total of 705 GO entries with aP-value < 0.05 were obtained,including 640 biological process (BP),31 cellular component (CC),and 34 molecular function(MF).The histogram of each category was shown in Figure 4.As for pathway analysis,85 targets participated in KEGG pathways with aP-value < 0.05,including TNF signaling pathway,IL-17 signaling pathway,Hepatitis B,Influenza A,Chagas disease(American trypanosomiasis),Pertussis,Kaposi's sarcoma-associated herpesvirus infection and so on.Among them,TNF signaling pathway mainly involved the genes PTGS2,Caspase 3 (CASP3),Caspase 8(CASP8),RELA,Mitogen-activated protein kinase 1(MAPK1) and IL-6; Genes associated with the IL-17 signaling pathway were PTGS2,Interleukin-4 (IL4),CASP3,CASP8,RELA,Mitogen-activated protein kinase (MAPK) and so on.And the top 20 pathways with smallP-values were presented as a bubble diagram in Figure 5.
Construction of “composition-targets-pathway”network
On the one hand,based on the top 20 critical key signaling pathways of HLJD for treatment of COVID-19,the targets involved mainly included: HSP90AA1,Adrenoceptor Beta 2,Checkpoint Kinase 1,Peroxisome Proliferator Activated Receptor Gamma and MAPK14,etc.On the other hand,these targets corresponded to 49 active compounds,including MOL000098-quercetin,MOL000173-wogonin,MOL002714-baicalein,MOL000422-kaempferol and MOL001689-acacetin and so on,demonstrating that various active compounds took effect through multiple targets and multiple signaling pathways.As shown in Figure 6,the circle,arrow and triangle nodes represent the compound,the target and the pathway,respectively.The bigger the shape is,the more significant the role it has played.In addition,the edges represent the active compound-target,signaling pathway-target and active compound-signaling pathway interactions,respectively.
Feasibility verification of molecular docking
At first,the complexes containing a covalent bond between the protein and ligand were removed,and all the docking calculations were performed in accordance with the set parameters.Next,the root-mean-square deviation (RMSD) between re-docking pose and original ligand conformation was calculated for evaluation.Eventually,the results indicated that RMSD was less than 0.2 nm during the simulation,demonstrating that the docking algorithm could reproduce the binding mode of receptor-ligand.Thus,the molecular docking method is computationally feasible and accurate.

Figure 4 GO enrichment analysis of treating COVID-19 targets with HLJD.(BP,Biological Process; CC,Cell Component; MF,Molecular Function)
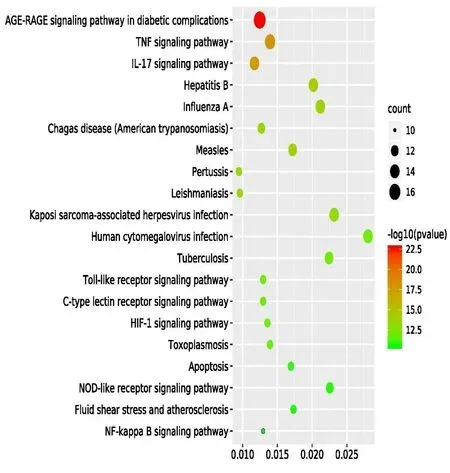
Figure 5 KEGG pathway analysis in treating COVID-19 targets with HLJD

Figure 6 “Composition-targets- Pathway” network of HLJD for treatment of COVID-19

Table 1 Docking results from SARS-CoV-2 3CL hydrolase and active ingredients with the currently effective drugs in HLJD
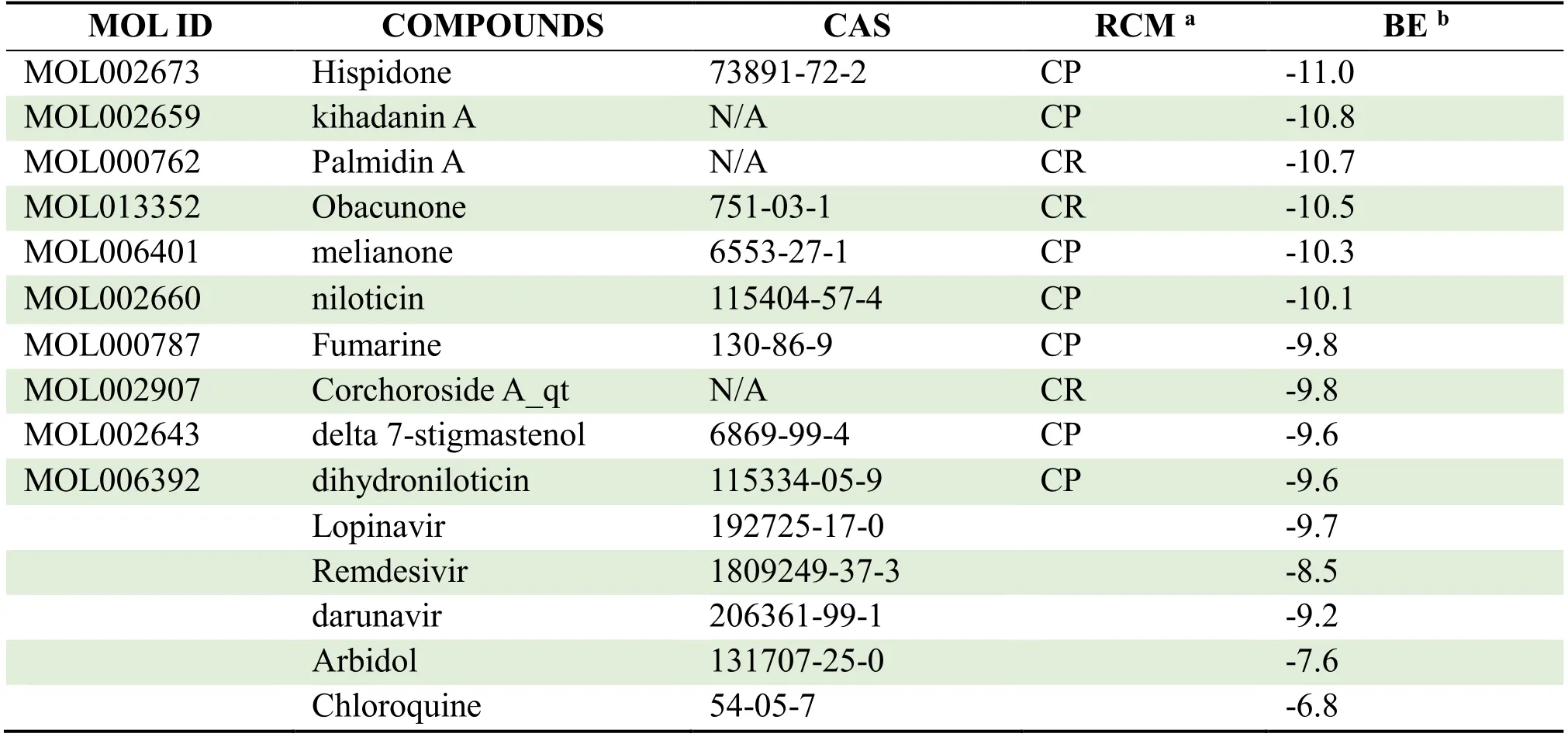
Table 2 Docking results from Spike glycoprotein and active ingredients with the currently effective drugs in HLJD

Table 3 Docking results from ACE2 and active ingredients with the currently effective drugs in HLJD
Virtual screening and molecular docking
The receptor-ligand set was considered stable when the binding free energy was lower than -7.0 kcal/mol,which indicated a stronger binding activity [19].The results of molecular docking were ranked from low to high in Table 1-3.As indicated,there were 64,94 and 76 compounds in HLJD whose binding energies with SARS-CoV-2 3CL hydrolase,Spike glycoprotein and ACE2 were all less than -7.0 kcal/mol,respectively.It was found that kihadanin A,Palmidin A,Hispidone and Obacunone bound with SARS-CoV-2 3CL hydrolase,Spike glycoprotein and ACE2 with high affinities.The molecular docking diagram of receptor proteins and a two-dimensional graph of interaction between receptor proteins and core active compounds in HLJD are shown in Figure 7 and Figure 8.

Figure 7 Molecular docking diagram of receptor protein with core active ingredients in HLJD
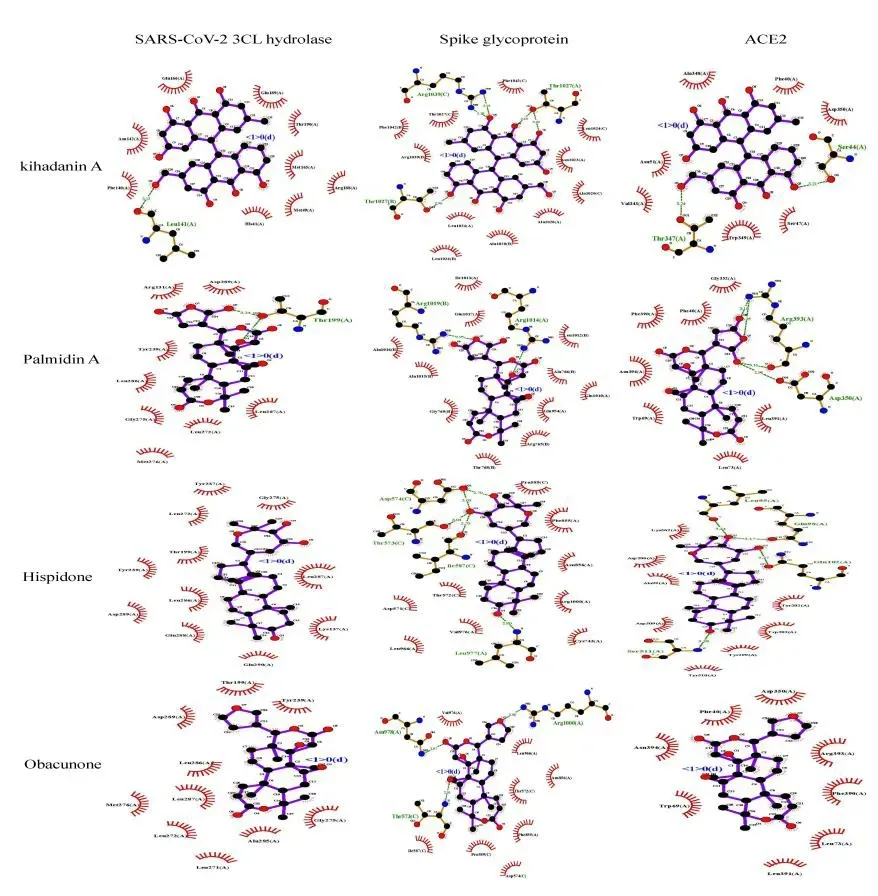
Figure 8 Two-dimensional graph of interaction between receptor protein with core active ingredients in HLJD

Figure 9 Molecular dynamics simulation RMSD of kihadanin A and SARS-CoV-2 3CL hydrolase

Figure 10 Molecular dynamics simulation RMSF of kihadanin A and SARS-CoV-2 3CL hydrolase
Molecular dynamics simulation results of complexes
After performing 50 ns of classical molecular dynamics simulations,RMSD of the trajectory was calculated to check the stability of the systems [20].As shown in Figure 9,it could be seen that the systems reached an equilibrium state after 20 ns whose RMSD values of all domains remained basically constant.These RMSD values fluctuated within 0.2 nm until the end of the simulations,indicating the stability of the complex systems.The root mean square fluctuations (RMSF) of the amino acid backbone atoms of the protein for all complexes were calculated to analyze the fluctuations of amino acids in complexes residue of Kihadanin A and SARS-CoV-2 3CL hydrolase during simulation.It was observed that the fluctuations of Kihadanin A and SARS-CoV-2 3CL hydrolase toward amino acids in 77-117,120-141,156-187,191-216,and 280-299 were relatively low (RMSF < 0.2 nm).The amino acids with less fluctuation in RMSF were consistent with the key amino acids,which might be due to the formation of stable hydrogen bonds,hydrophobic interaction or π-π stacking between the complexes and the proteins.In this way,stable protein complexes were formed,making the key amino acids more stable,and contributing to the interaction between receptor and ligand,as shown in Figure 10.
Analysis of total binding free energy of MM-PBSA
The binding free energy consists of three parts: the molecular mechanics energy (MM),including van der Waals contributions in the gas phase,and Coulomb electrostatic interaction energy; polar solvation energy(PB); nonpolar solvation energy (SA).Next,binding free energy calculations were performed to evaluate the stability associated with the formation of the receptorligand complexes.Van der Waals interactions,Coulomb electrostatic interactions,PB energy and SA energy between the Kihadanin A and SARS-CoV-2 3CL hydrolase were -119.537,-25.779,74.5035 kcal/mol and -11.821 kcal/mol,respectively,which implied that the van der Waals forces would usually be dominant in the interactions between Kihadanin A and SARS-CoV-2 3CL hydrolase,followed by electrostatic interactions and SA energy.In contrast,PB energy hindered the binding between Kihadanin A and SARSCoV-2 3CL hydrolase.The results were depicted in Table 4.
Decomposition of binding free energy of MM-PBSA
In order to identify the key amino acids involved in the binding between Kihadanin A and SARS-CoV-2 3CL hydrolase,the total binding free energy was decomposed to single amino acid residues by using the MM-PBSA method.The results showed nine residues contributed a lot to the total binding free energy,including Asp289,Leu287,Leu286,Met276,Let272,Tyr239,Thr199,Gly275 and Arg131.Except that PB energy served as the main mode of action between Arg131 and Kihadanin A,other residues interacted with Kihadanin A mainly through the electrostatic interactions and Van der Waals interactions,as shown in Table 5.

Table 4 Binding free energy of kihadanin A and SARS-CoV-2 3CL hydrolase (kcal/mol)
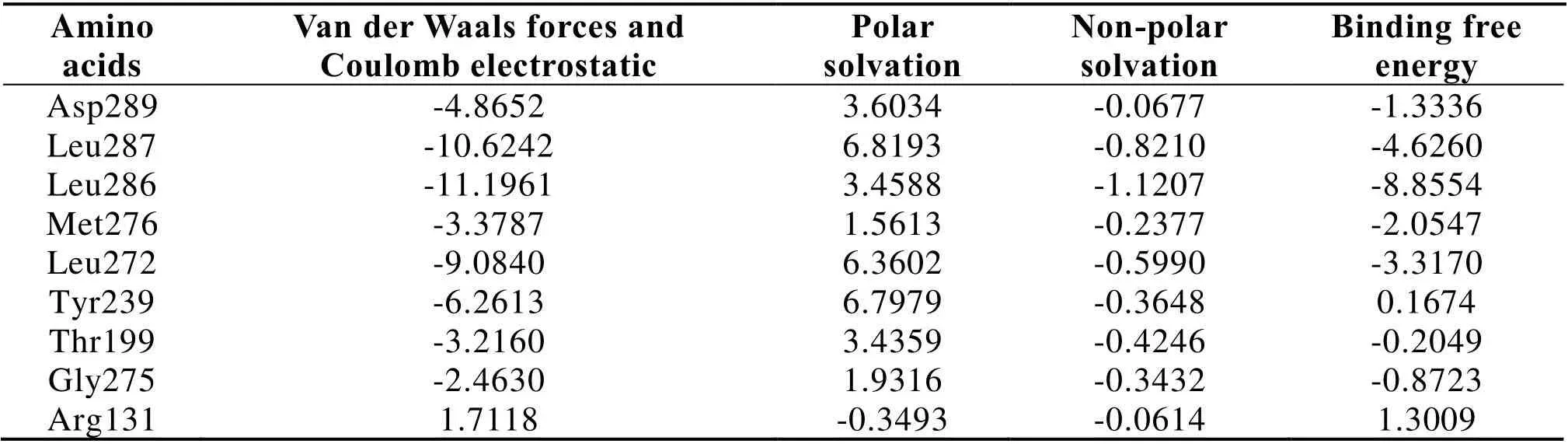
Table 5 Binding free energy decomposition of kihadanin A and SARS-CoV-2 3CL hydrolase (kcal/mol)
Discussion
Most infectious diseases caused by viruses fell under the category of “plagues” in TCM,which are mostly the results of the invasion of evil energy or the lack of vital energy in the body.COVID-19 is a kind of epidemic disease in TCM,and the evil energy of epidemic toxin is the main pathogenic factor.On the basis of the TCM syndrome differentiation,the clinical manifestations in patients mainly include pulmonary obstruction from epidemic toxin accompanied by yellow moss and high fever.In this case,the method of lung-clearing and detoxification could be adopted to detoxify the lung and dissipate heat [22].Heat-clearing and detoxifying drugs are mainly used in the fight against viruses,represented by HLJD.
The “materials-compounds-targets” network analysis showed: for one thing,prostaglandinendoperoxide syntheses (including PTGS1 and PTGS2)played an important role in the arachidonic acid’s metabolic pathway,inhibiting the generation of PTGS2,and consequently reducing the inflammatory response.For another,HSP90AA1 was associated with immunity to elicit antitumor immune responses [23].Following the topological screening in the PPI network,core targets including CASP8,IL-6,MAPK1,MAPK14 and RELA were optimally obtained.The CASP8 mediated apoptosis between different cell death patterns,serving as a molecular switch that controlled apoptosis,necrosis and pyroptosis [24].After preliminary clinical observation,critically ill patients with COVID-19 might exhibit a significant increase in inflammatory cytokines such as IL-6 and TNF.IL-6 could contribute to the generation of immune cells by activating the Jak/Stat signaling pathway,which played an important role in the development of primary and secondary cytokine storms [25]; MAPK worked as a regulator in many cellular processes including cell proliferation,differentiation,apoptosis,cells’ stress stimuli to inflammatory response and virus infection status.And simultaneously,CR,and FG played the role of heatclearing and detoxification by inhibiting the MAPK pathway [26].
According to KFGG analyses,two cancer-related pathways,including “non-small-cell lung cancer pathway” and “small-cell lung cancer pathway”,both involved the expressions of RB Transcriptional Corepressor 1 (RB1) and BCL2 Associated X (BAX)genes.The four virus-related pathways revealed the expressions of BAX,CASP3,and RELA genes,which were all the therapeutic targets of main ingredients like baicalein,wogonin,kaempfero and quercetin.Meanwhile,previous studies also found that CR extract was a potential antiviral drug [27].Berberine has the power to inhibit influenza-induced viral pneumonia; in addition to inhibiting apoptosis and exerting the antitumor activity,wogonin,acacetin and beta-sitosterol also have the potency of anti-oxidation,inflammation reduction and bacterial infection inhibition; moreover,baicalein and wogonin could also significantly ameliorate influenza A virus-induced acute lung injury[28].
Forty-nine active compounds in HLJD were identified in the results of the “active compoundstargets-pathways” network,which exerted the curative effect on 53 targets,including HSP90AA1,Adrenoceptor Beta 2,Checkpoint Kinase 1,Peroxisome Proliferator Activated Receptor Gamma and MAPK14.They participated in multiple essential biological processes,such as immunity and inflammation,oxidative stress,cell proliferation,differentiation and apoptosis,got involved in the TNF and IL-17 signaling pathways,and regulated the immune-inflammatory factors.In addition,they also played an important role in inhibiting the inflammatory response,regulating apoptosis and exerting the antiviral effect.HENG et al.[29] conducted target predictions for active compounds in HLJD,so as to investigate its network pharmacology mechanism on the inflammation of macrophages and conjecture its regulatory role in treating inflammation through sphingolipid and glutamine metabolism.According to the findings of LIU et al.[30],the effective components of HLJD had the capacity to reduce the intracellular Lipopolysaccharide,thus suppressing the expression of Caspase-11,reducing apoptosis and facilitating the treatment of sepsis.
The common compounds of HLJD are listed based on the structure-based virtual screening and molecular docking results,and these compounds include epiberberine,berberine,worenine,berberrubine,coptisine,β-sitosterol,quercetin and stigmasterol.Among them,berberine (-7.8 kcal/mol),quercetin (-7.7 kcal/mol),epiberberine (-7.7 kcal/mol) show good affinity with SARS-CoV-2 3CL hydrolase; while coptisine (-9.2 kcal/mol),worenine (-9.0 kcal/mol),and berberine (-8.6 kcal/mol) bind well to Spike glycoprotein with excellent affinity.In addition,coptisine (-8.8 kcal/mol),stigmasterol (-8.4 kcal/mol),and berberrubine (-8.4 kcal/mol) exhibit good affinity with ACE2.It can be concluded that low conformational energy indicates high affinity between common compounds and relevant target proteins,which have similar binding energies to Lopinavir,Ritonavir,Arbidol and Chloroquine.Moreover,as the main constituent of Phellodendri Chinrnsis Cortex,Obacunone can be used to effectively suppress bleomycin-induced lung injury and pulmonary fibrosis in mice,and regulate the expressions of inflammatory lymphokines in bleomycin-induced lung injury tissues.In summary,Obacunone not only serves as an effective component for preventing and treating lung injury and pulmonary fibrosis,but also has low toxicity and little influence on liver and kidney [31]; thus it is important for clinical practice.However,since some compounds identified in HLJD are not common and rarely studied,further studies shall be conducted to make better use of such compounds.
The main driving forces in the binding reaction of Kihadanin A and SARS-CoV-2 3CL hydrolase were van der Waals forces.Coulomb electrostatic interactions and SA energy had little effect on binding,while the PB energy impeded their interaction.The hydropathy plot suggested that naringin revealed hydrogen bonding with residues,such as Asp289,Leu287,Leu286,Met276,Let272,Tyr239,Thr199,Gly275 andArg131,forming the same hydrophobic pocket.These findings might provide some reference for the development of SARS-CoV-2 3CL hydrolase inhibitor as well as the structure-activity relationship of TCM compounds.
To sum up,researchers have not gained a comprehensive understanding of the pathogenic mechanism of COVID-19 up to now.Besides,the computational chemical and biological methods are still not accurate enough; the action targets and structures of chemical ingredients in TCM have not been fully explored.Therefore,the determinations of Kihadanin A,Palmidin A,Obacunone,Hispidone and other rare compounds are extremely important for the pharmacological study and quality control.Additional pharmacodynamic and clinical studies shall also be carried out for further verification,thus providing new ideas for clinical diagnosis and treatment of COVID-19.
Conclusion
In this paper,we preliminarily explored the potential therapeutic mechanism for HLJD to against COVID-19 and further verify the combination mode.We hope that the results will help to further study on the active ingredients and mechanism of HLJD for anti-COVID-19.
杂志排行

TMR Modern Herbal Medicine的其它文章
- The therapeutic efficiency of ursodeoxycholic acid on Gilbert syndrome complicated by aplastic anemia: a series of case reports
- Speech on the evidence-based traditional Chinese medicine research 20th anniversary conference
- Study on the mechanism of Fructus Forsythiae and Folium Nelumbinis on acute pharyngitis based on network pharmacology
- Shiqi herbal tea reduces the susceptibility to H1N1 influenza virus in stressed mice.
- A systematic review on the neuropharmacological activities of Oligosaccharide ester in Polygalae Radix
- Uncovering the multi-target pharmacological mechanism of Xuebijing injection against sepsis by a systems pharmacology approach