钨/铜界面处氢原子与空位相互作用的第一性原理计算
2021-01-21柳文波王东杰张朋波
柳文波,何 欢,王东杰,张朋波
(1.西安交通大学 核科学与技术学院,陕西 西安 710049;2.西安交通大学 陕西省先进核能工程研究中心 陕西省先进核能技术重点实验室,陕西 西安 710049;3.中国原子能科学研究院,北京 102413;4.大连海事大学 理学院,辽宁 大连 116026)
随着社会生产力的发展,人类对能源的需求日益增加。核聚变能源具有效率高、经济性好、清洁绿色等特点,是一种未来重要的清洁能源。目前,聚变堆研究已成为各国能源发展中的重中之重,我国也正在全力发展中国聚变工程示范试验堆(CFETR)[1-2]。偏滤器作为CFETR工程装置中最核心的部件之一,直接面对高温等离子体和强中子辐照环境,承担着排除高通量能量密度和粒子流的功能,服役环境相当恶劣,在CFETR设计的第1阶段中,其将承受10 MW/m2的峰值热流密度[3]。而对于CFETR偏滤器,现多采用穿管型的结构,该结构采用第一壁材料模块钨,焊接在CuCrZr热沉/冷却水管上,中间衬有铜过渡层。这样的结构设计直接导致了CFETR偏滤器采用的穿管型结构的钨/铜异质材料的连接界面将成为氢同位素渗透滞留的高速通道和捕获陷阱[4]。氢原子会更易在界面处渗透滞留,造成偏滤器部件热力学性能的退化。因此,研究钨/铜界面中氢原子与点缺陷的行为有助于理解偏滤器在CFETR中的工作原理及提升其抗辐照性能[5]。
相对于钨中大量的实验[6-7]和模拟[8]研究,钨/铜界面,尤其是界面中氢原子的滞留行为研究较少。Girault等[9]通过X射线衍射实验发现,钨/铜界面中,钨原子层主要为{110}晶面,而铜原子层主要为{111}晶面。Liang等[10]对钨/铜界面结构稳定性进行了第一性原理研究,通过改变外延层和衬底层的表面方向及层数,(111)Cu/(110)W和(110)Cu/(110)W被认为是最易形成的界面类型。Ma等[11]对氢原子在钨/铜界面行为同样进行了第一性原理研究,发现氢原子更倾向于扩散至钨/铜表面和铜晶格中。然而,对于钨/铜界面中氢原子的行为研究目前仅局限于单个氢原子行为,对于氢原子与缺陷的相互作用机理尚不清楚。因此,本文将通过第一性原理对(111)Cu/(110)W界面中氢原子行为及其与点缺陷的作用进行研究。
1 模型建立
本文所采用的是广泛应用于材料计算领域的第一性原理方法。基于密度泛函理论的第一性原理方法,能从原子和电子尺度上精确地理解缺陷的行为机理,是目前研究微观缺陷的最有效研究手段之一。所用的软件为基于平面波方法的VASP软件[12],在整个弛豫及结构优化的计算过程中采用基于广义梯度近似(GGA)方法,泛函为PBE泛函;描述体系的电子相互交换能量,平面波截断能设置为500 eV,能量及力的判据分别为10-5eV及108eV/m,K点设置为3×3×1。
首先基于优化后的钨(晶格常数为3.17×10-10m)、铜晶格(晶格常数为3.63×10-10m),切面得到(111)Cu、(110)W表面,表面能计算结果分别为1.30 J/m2、3.06 J/m2,与已研究结果(铜:1.387 J/m2[13],1.275 J/m2[10];钨:3.348 J/m2[10])相符合。因此,根据构建的表面模型及Liang等[10]对钨/铜界面结构稳定性的研究,建立图1所示的钨/铜界面模型。其中钨作为衬底层,铜作为沉积层,原子层厚度分别为6层和4层,真空层的厚度为15×10-10m。整个体系包含90个原子(54个钨原子,36个铜原子)。由于在实际的穿管部件制备中,钨材料一般作为钨/铜界面的衬底层,铜材料是在钨衬底上生长,因此晶格常数选取的是下层钨表面的晶格常数。晶矢a、b、c分别为(8.22×10-10m,0 m,0 m)、(2.74×10-10m,7.75×10-10m,0 m)和(0 m,0 m,35.26×10-10m),晶胞内晶格的夹角α、β、γ分别为90°、90°和70.5°。计算过程中只固定最底层的原子,以模拟真实的界面情形。

图1 钨/铜界面结构Fig.1 Structure of W/Cu interface
2 模拟结果分析
2.1 界面处氢原子的稳定位点
本文研究单个氢原子在钨/铜界面附近的滞留行为,以获取氢原子在界面处的稳定位置。由于氢原子的原子半径远小于钨原子和铜原子,因此氢原子在面心立方或体心立方金属中一般会占据两个高对称点位置,即八面体间隙位(OIS)和四面体间隙位(TIS)。根据钨/铜界面中不同高度的高对称点位置,考虑6种高对称点位置的氢原子,如图2a所示。通过第一性原理结构优化,结果发现在有些高对称点位置上,氢原子是无法稳定存在的,优化后氢原子的位置如图2b所示。
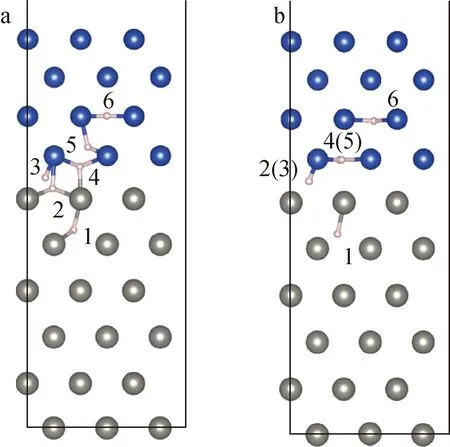
a——结构优化前;b——结构优化后图2 钨/铜界面中氢原子位置示意图 Fig.2 Schematic of hydrogen atom position in W/Cu interface
通过比较不同位置氢原子下优化后体系的能量,发现2(3)(Hmiddle)和4(5)(Hupper)是钨/铜界面中氢原子最稳定的位置,而其他位置是氢原子亚稳定位置。该结果与Ma等[11]的计算结果是一致的,这表明氢原子在钨/铜界面中更倾向于存在界面中或铜晶格中,而不是钨晶格中。根据钨/铜界面的几何模型,该现象可能是由于钨/铜界面处的原子空间较大及铜的晶格常数相对较大导致的。
2.2 界面处的空位
在强中子辐照下,材料内部将产生大量的缺陷,缺陷的演化会影响材料的力学性能,最终导致部件材料失效。因此,研究钨/铜界面中缺陷的存在有助于在原子尺度上理解材料的力学性能变化。通过在钨/铜界面中引入不同位置的空位和间隙发现,由于钨原子和铜原子的原子半径较大,两者的间隙均很难稳定地存在于界面中。因此,以下的研究中仅考虑空位,而不考虑间隙。根据图1中钨/铜界面模型,本文将界面模型中不同层数中的单个晶格原子移去,以获得不同层数下的单空位模型。
1) 铜空位
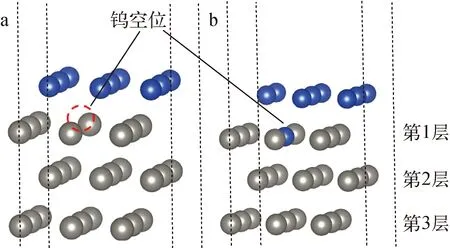
在钨/铜界面中,分别将4层外延铜原子层各移去1个铜原子后进行结构优化。计算结果表明,靠近钨/铜界面的铜空位,即第1、2、3层,是不稳定的,这3层的铜空位会自发地迁移到最上层的铜原子层,最终形成空位的迁移过程。该现象说明铜空位不能单独在钨/铜界面处稳定存在,会自发地迁移到上表面,形成上表面的空位形态,这种不稳定性与纯铜中空位能稳定存在的现象是完全不同的。图3为钨/铜界面中铜空位位置示意图。
2) 钨空位
相比于铜空位在钨/铜界面中的不稳定性质,本文发现钨空位在钨/铜界面中是一种相对稳定的缺陷。结合不同原子层中钨空位的结构优化后结果,钨空位只有在最靠近钨/铜界面的原子层时是不稳定的,而第2层及第2层以上原子层中的钨空位都是能稳定存在的(图4)。对于稳定的钨空位,其形成能为4 eV左右,该能量要略大于纯钨中的空位(3.2~3.5 eV)[14],这表明相比于纯钨,钨/铜界面更难形成钨空位。
a——结构优化前;b——结构优化后图4 钨/铜界面中钨空位位置示意图Fig.4 Schematic of W vacancy position in W/Cu interface
对于最靠近钨/铜界面的原子层钨空位,其不稳定原因主要是钨空位的存在为上层铜晶格中的原子提供了1个可移动的位置,半径小的铜原子会穿过整个界面,占据钨空位的位置,形成1个铜反位。与此同时,该铜原子在铜晶格中留下1个铜空位的存在。
由以上结果可知,对于钨/铜界面中的空位,铜空位很难稳定存在于界面附近,其更倾向于远离界面,而钨空位更易稳定存在于界面附近,只有最靠近界面原子层的钨空位会吸引上层的铜原子,形成铜反位。
2.3 界面处氢原子与空位的相互作用
在实际的CFETR工作环境中,由于聚变堆燃料的特殊性,材料内部会出现大量的氢原子。在强中子辐照下,材料内部将不可避免地产生大量点缺陷。因此,研究界面处氢原子与点缺陷相互作用有利于理解偏滤器在接近实际工况下的工作原理。针对钨/铜界面中最稳定位置的两种氢原子Hmiddle与Hupper,分别假设9种不同位置处的空位以探究界面处氢原子与点缺陷的相互作用,图5为钨/铜界面中氢原子与空位的相对位置示意图。
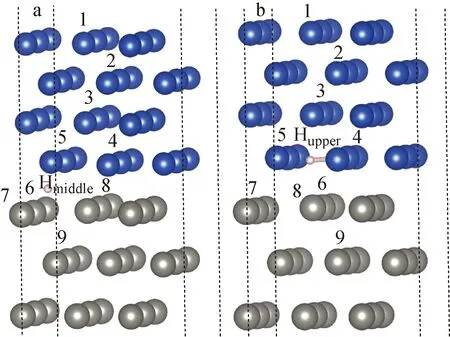
a——Hmiddle;b——Hupper图5 钨/铜界面中氢原子与空位的相对位置示意图Fig.5 Schematic of relative position of hydrogen atom and vacancy in W/Cu interface
在氢原子周围分别引入不同的单空位,钨/铜界面中氢原子在空位存在下的移动距离列于表1。由表1可知,大多数位置的空位对氢原子的影响较小,氢原子只有微小的向空位位置移动靠近的现象出现。对于Hmiddle原子,虽然这种氢原子在c方向上与铜、钨晶格距离相同,但其在铜空位存在时,移动距离(<0.2×10-10m)较小,而在有钨空位存在时,特别是对于最上层钨空位,Hmiddle原子移动了较大的距离。而Hupper原子则与此相反,虽然这种氢原子处在铜晶格中,但其在铜或钨空位存在时,均会移动较大的距离。这种现象说明相比于铜空位,钨空位更能吸引界面中间的原子,Hmiddle原子更加稳定,移动的距离整体较少。值得注意的是,对于Hmiddle中的空位7及Hupper中的空位6,均出现了氢原子移动到铜或钨晶格原子上的现象,这进一步说明钨空位吸引氢原子的能力更强。
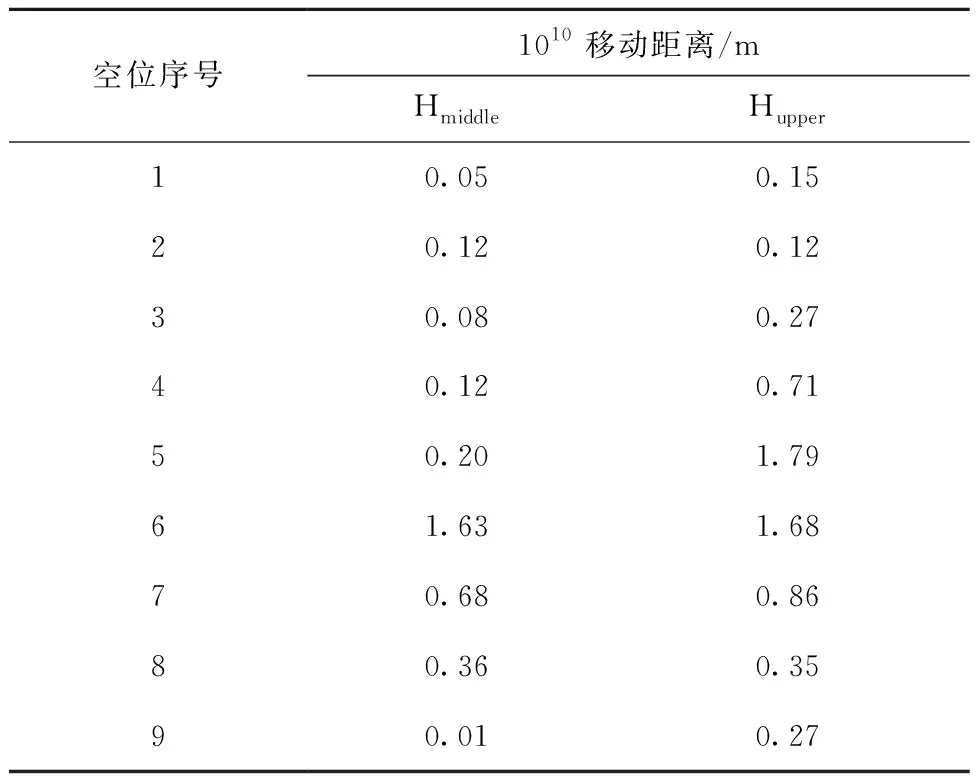
表1 钨/铜界面中氢原子在空位存在下的移动距离Table 1 Displacement distance of hydrogen atom in W/Cu interface with vacancy
与此同时,对于铜空位2、3和4,在Hmiddle或Hupper存在的情况下,3种空位并未出现铜空位会移动到上表面的现象,这表明氢原子在界面处存在时可阻碍空位向上表面迁移,从而将空位留在界面附近。这种空位的存在导致氢原子更易滞留在界面处,从而可能导致氢泡的形成。
相比于纯钨中钨空位对氢原子的强吸引力[15-16],本文钨/铜界面中钨空位对氢原子的研究结果发现,只有钨空位7出现了氢原子进入空位的现象,这表明界面的存在降低了钨空位吸引氢原子的能力。
通过研究不同位置的空位与氢原子的相互作用,发现当界面处没有空位时,氢原子更倾向于占据界面位置及铜晶格内部;而当界面处存在空位时,氢原子更倾向于占据钨空位。与此同时,界面处的氢原子会阻止空位迁出界面,从而在界面处形成能滞留多个氢原子的空位。
3 结论
本工作利用第一性原理方法研究了钨/铜界面中氢原子与点缺陷的相互作用,研究结果如下。
1) 钨/铜体系中的氢原子更倾向于在钨/铜界面处及铜晶格中稳定存在。
2) 相比于纯金属体系,界面附近的铜空位十分不稳定,它会自发地迁移到上表面,而大部分钨空位能稳定存在,靠近钨/铜界面的钨空位还会吸引铜原子。
3) 两种稳定位置的氢原子与不同位置空位的相互作用的模拟结果表明:在空位的影响下,氢原子会产生一定的移动距离,钨空位对氢原子的吸引能力比铜空位对氢原子的吸引能力强。氢原子的存在会抑制铜空位向远离界面方向的迁移,界面附近氢原子与空位的共同存在可能是导致钨/铜界面处形成氢气泡的主要原因。
