土壤病毒的研究进展与挑战*
2021-01-05王光华刘俊杰朱永官
王光华,刘俊杰,朱 冬,叶 茂,朱永官,4
(1. 中国科学院东北地理与农业生态研究所/中国科学院黑土区农业生态重点实验室,哈尔滨 150081;2. 中国科学院生态环境研究中心,北京 100085;3. 中国科学院南京土壤研究所/中国科学院土壤环境与污染修复重点实验室,南京 210008;4. 中国科学院城市环境研究所,厦门 361021)
病毒是个体非常微小、结构简单,由蛋白质外壳包被的内部含有遗传物质核酸的非细胞型生物实体。按照核酸的组成,病毒可划分为双链DNA(dsDNA)、单链DNA(ssDNA)、双链RNA(dsRNA)、正单链RNA(ssRNA+)和负单链RNA(ssRNA-)。通常病毒被认为是不具有酶系统,不能够独立繁殖生活,病毒的繁殖必须依赖于寄主系统,所以长期以来病毒是否被视为生物一直存在争议[1]。但这种现象目前有所改变,科学家们从多种环境中发现了一些感染原生动物的病毒粒子,其尺寸较大,被称为巨大病毒,如梅格病毒(Megavirus)和潘多拉病毒(Pandoravirus)粒子直径分别达到440 nm和1 000 nm,其基因组大小分别为1.26 Mbp和2.5 Mbp,这些巨大病毒具有独特的酶系统[2-4],有的病毒还被其他病毒(噬病毒体Virophage)侵染[2,5],这些发现动摇了学术界对生命之树的认知,有观点认为病毒可看做为生命的第四个域——病毒域[6]。事实上,病毒可以侵染生命之树上其他三域的生物,通常学术界将侵染细菌和古菌的病毒,即侵染原核微生物的病毒称为噬菌体。目前国际上分离获得的细菌病毒有6 000多株,而古菌病毒仅100多株[7]。与细菌病毒遗传物质组成不同,古菌病毒的遗传物质多是dsDNA,少数是ssDNA,尚未发现含有RNA的古菌病毒[8]。有观点认为病毒可能较地球上所有生物的共同祖先露卡(LUCA)出现的更早[9],地球上所有具有细胞结构的生物都能够被一种甚至多种病毒所侵染[10],病毒在侵染过程中基因不断地突变,也不断地与寄主间进行遗传物质的交流,从而确保病毒能够生存下去,也使寄主生物获得新的基因,产生新的性状,促进了地球生物的进化演替。
土壤是各种生物的重要栖息地,土壤中存在着数量巨大,种类繁多的病毒生命体。虽然学术界普遍意识到病毒在土壤各种生态过程中可能起到重要的作用,但由于受到土壤的异质性、多样性,以及研究手段等方面的限制,目前国际上对土壤病毒研究的重视程度远落后于流动的海洋环境[11-13]。本文从生态学角度,基于目前国内外病毒研究现状,尤其是针对土壤病毒的研究给予回顾,期望起到抛砖引玉的作用,引起国内同行对土壤病毒研究的关注和重视。
1 土壤病毒形态与数量
1.1 土壤病毒的形态
病毒大小通常是纳米级的,普通光学显微镜无法捕捉到病毒的身影,必须通过透射电子显微镜(TEM)才能观测到。病毒形态多种多样,目前发现古菌病毒形态有16种,而细菌病毒形态有9种,可见虽然目前得到的古菌病毒纯分离株远少于细菌病毒,但其形态多样性却高于细菌病毒[7]。土壤中的古菌病毒和细菌病毒多以具有头尾结构的有尾噬菌体为主。有尾噬菌体是dsDNA病毒,按其尾部形态特征可划分为短尾(Podophage)、长尾(Siphophage)和肌尾(Myophage)噬菌体[7]。土壤中其他病毒形态还有丝状、杆状、棒状、瓶状、水滴状、球状和二十面体状等[8]。真核生物病毒没有头尾结构,形态更加多样,有的蛋白质外壳上还有修饰成分,与原核生物病毒有很大的不同(图1)。

图1 侵染细菌、古菌和真核生物的dsDNA病毒分类及形态示意图(改自Prangishvili等[14])Fig. 1 Classification and morphological diagrams of the dsDNA viruses that infect bacteria,archaea and eukaryotes(Modified from Parangishvili et al. [14])
1.2 土壤病毒存在形式
病毒在土壤中的存在形式大致可归为3种:少部分病毒以游离状态存在于土壤溶液中;大部分病毒被吸附在土壤颗粒上;还有一部分是温和性噬菌体(lysogenic or temperate phage),其基因组插入在寄主基因组上,以溶原(prophage)状态存在于细胞内[11,15]。温和性噬菌体在土壤中普遍存在,尤其是在寄主分布不均匀[16]、寄主数量少[17-18]或寄主细胞生长状况不好[17,19]的情况下,噬菌体以溶原状态伴随着寄主细胞繁殖而繁殖,避免受到外面不利环境条件的影响,从而确保噬菌体在寄主种群中能够长期存活繁殖下去的最佳方式。一般而言,在某个环境中病毒与细菌比值(VBR)数值小时,噬菌体主要以溶原状态存在,而当VBR数值大时,则以烈性噬菌体(lytic phages)为主[20-22]。相比于营养贫乏的海水环境,营养丰富的土壤中溶原性噬菌体占比较高[20,23]。溶原性噬菌体也是极端干热沙漠条件下噬菌体主要存在形式[24],而在冷沙漠中噬菌体主要以游离的烈性噬菌体为主[21]。溶原状态噬菌体在土壤中存在,也可通过土壤细菌全基因组测序而发现,有研究表明许多细菌基因组中含有1个或多个前噬菌体元件(prophage element)[25],其中默冗子(moron)是前噬菌体元件的一个重要部分[26-27]。默冗子在噬菌体基因组中不具有编码功能,但当它整合在细菌基因组中则通过基因溶原性转换(lysogenic conversion),使细菌获得新的性状和功能,提高细菌环境生存适应能力[28-30]。
1.3 土壤病毒数量
目前地球生物圈上到底有多少病毒还没有准确数据,普遍认为全球病毒数量>1031[31]。有学者依据不同生境中病毒与细菌或与微生物的比值(VBR或VMR),推算出全球病毒粒子的数量为4.80×1031,其中在沉积物和土壤中数量分别占到87%和10%,而在海洋水体中只占2.7%,可见土壤环境中病毒数量非常巨大[32]。不同环境中病毒丰度存在较大差异,将病毒颗粒(virus-like particles,VLPs)用荧光核酸染料染色,然后在荧光显微镜(EFM)下计数发现,每毫升海水中VLPs在105~107个之间[33-34],河口或湖泊淡水中VLPs可达到每毫升108个[35];而在森林土壤和农田土壤中,每克土壤VLPs数量分别在1.31×109~4.17×109个和0.87×109~1.1×109个之间[36]。最近,Williamson等[37]对热带沙漠、寒带沙漠、森林、农田和湿地等环境下的土壤病毒和细菌数量进行Meta数据分析发现,病毒在沙漠土壤中丰度最低,森林土壤中丰度最高,土壤类型与病毒数量高度相关(r=0.803,P<0.001);发现土壤病毒数量与土壤细菌数量呈高度正相关(r=0.647,P<0.001);发现土壤病毒数量与土壤pH呈显著负相关(r= -0.352,P=0.009)。同时他们也认为土壤温度和湿度也是影响土壤病毒数量的重要土壤因子,但由于数据的缺乏,两者之间的相关性尚未建立起来[37]。需要指出的是,目前对土壤病毒数量的研究工作主要是针对土壤中游离的和被土壤颗粒吸附的病毒,尚无法大规模地开展针对土壤中溶原性病毒的计数研究,所以现有观察到的数据可能远低于土壤中病毒的真实数量。
由于大部分病毒被吸附在土壤颗粒上,如何从土壤中将病毒高效率地分离提取出来是准确测定病毒数量的前提条件。美国学者Wommack团队[38]对比研究了10%的牛肉膏溶液、250 mmol·L-1甘氨酸溶液、10 mmol·L-1焦磷酸钠溶液和1%柠檬酸钾溶液从粉砂壤土和砂壤土中提取病毒的效果,发现1%柠檬酸钾溶液是最佳的土壤病毒浸提剂。在此基础上,Trubl等[39]改进了提取剂配方,发现1%柠檬酸钾溶液+10%磷酸盐缓冲液+5 mmol·L-1的EDTA溶液+150 mmol·L-1硫酸镁溶液对富含有机质的泥炭和沼泽土壤中病毒提取效率最高,提取出的病毒数量是其他提取剂的2倍。而张辉等[40]以MS2和φx174作为指示病毒,研究发现含有0.04 mol·L-1焦磷酸钠的3%牛肉膏溶液提取效果最好,用此提取剂指示病毒在红壤土、红黏土和潮土上的回收率达63%~98%,但在黄泥土上的回收率仅30%。可见同一种病毒提取剂的提取效率因土壤类型而异。需要注意的是,从土壤中提取病毒溶液的操作过程多伴随采用超声波、涡旋、剧烈震荡,有的甚至采用珠打法(bead-beating)等物理分散技术,这些剧烈的分散过程可以导致有尾噬菌体尾部结构折断或丢失,从而降低了观察到的有尾噬菌体比例[41-42]。
VMR或VBR数值大小也能间接地反映出环境病毒数量的高低。VBR在不同环境中的变幅很大,如在海洋和湖泊水体中数值在3~25之间[33-34,43],而在稻田水中变幅在0.11~72之间[44]。与水体环境相比,土壤中VBR变幅更大,有研究发现森林和农田土壤中的VBR分别为10和3 000[36],而在沙漠土壤中其数值仅0.15~1.66[45]。在根际微域环境中,有研究发现每克小麦根际土壤中VLPs是1.18×109个,与非根际土壤病毒数量没有显著差异,从而导致根际VBR为0.27,而非根际土壤是4.68[41]。这一发现表明尽管根际土壤细菌数量非常大(根际效应),但根际细菌病毒并没有体现出同步的增加规律,即根际细菌病毒数量没有体现出根际效应。这种现象与通常认为的细菌-病毒生长模型“杀死优胜者Kill-the-Winner”不符合[34,46]。最近,来自国外科学家在对珊瑚礁后生动物黏膜表面细菌和病毒之间相互作用研究中发现一个新的细菌-病毒生长模型,即“搭乘优胜者Piggyback-the-Winner”[47]。在这个模型中,病毒以溶原状态将病毒基因整合到寄主细胞体内,随着寄主细胞繁殖而繁殖,避免与其他病毒竞争,也避免触动宿主自身的免疫系统。这种“搭乘优胜者”模型是否是植物根际细菌病毒的生存策略还未见直接证据支持。此外,也有学者认为根际细菌病毒数量与非根际土壤无差异的原因也可能与根际细菌受到根系分泌物选择的作用而不易于被病毒侵染有关[13]。
1.4 土壤病毒生物量
全球病毒数量保守估计在1031以上,那么这些病毒生物量是多少呢?Cobián Güemes等[32]根据一个典型噬菌体含有50 kbp的DNA质量为0.054 fg(飞克),而壳蛋白质质量为0.028 3 fg,推算出全球4.80×1031VLPs的质量是3.95×1015g(3.95 Pg或3.95 Gt)。按照典型病毒化学计量学C︰N︰P =20︰6︰1计算[48],全球病毒生物量估算有2.92 Pg C、0.88 Pg N和0.15 Pg P。如果全球原核生物含有350~500 Pg C[49],那么病毒生物量碳约占原核生物量碳的0.58%~0.83%。而另一篇文献认为全球所有生物只有大约550 Gt C,其中细菌为70 Gt C,古菌大约为7 Gt C,病毒含有大约0.2 Gt C[50],这一估算数值较Cobián Güemes等[32]报道数据缩小了10倍,但两篇文献均表明病毒生物量相对于原核生物生物量而言非常小。与细菌细胞C︰N︰P的化学计量为60︰16︰1相比[51],细菌病毒颗粒富含N和P,尤其是P元素。虽然病毒生物量相比原核生物和真核生物而言非常小,但其数量巨大,周转快,在全球生物地球化学循环中起到非常重要的作用[52]。以海洋环境为例,有研究估测每天有1028个细菌被病毒感染而裂解,其中大约30%是具有光合作用的蓝细菌,60%是异养细菌[52-53]。海洋中某些区域有超过5%的可溶性有机磷和氮来源于病毒颗粒[48]。有报道指出海洋环境中大约6%~25%的碳循环量是由病毒驱动的[33],但在土壤环境中,病毒是如何及在多大程度上驱动碳循环过程还鲜见报道。
2 土壤病毒多样性
由于病毒不能独立繁殖,而大量环境微生物的不可培养性,无疑限制了对病毒多样性的认知。全球到底有多少种病毒尚无确切的答案。现有的报道均为基于推测的结果,例如有文献报道[54-55],全球有1 000万种微生物(主要是细菌和古菌),而每种微生物可被10种不同的病毒侵染,则全球会有超过1亿种的病毒[56-57]。笔者认为这种推测有可能高估了病毒的多样性,因为随着研究的不断深入,发现病毒与被侵染寄主之间具有很强专一性的观点被不断修正[58],如有些细菌病毒在细菌分类属、目,甚至在门水平上能够跨物种感染不同寄主[59-60]。最近,Williamson 等[37]基于病毒宏基因组数据分析发现土壤中病毒基因型(genotype)在1 000~1 000 000之间;与土壤相对应,海洋中病毒基因型在532~129 000之间,在淡水环境为400~40 000之间[61]。可见土壤病毒多样性的上限和下限均高于水体环境中的病毒,充分说明土壤病毒是一个巨大的未知基因资源库。
2.1 分子标记基因解析病毒多样性
采用保守性的引物,利用PCR技术对原核微生物的16S或真核生物18S rRNA基因片段进行扩增,然后对PCR产物进行基因信息解析,是目前研究环境微生物基因多样性和群落组成的主流技术。然而对于病毒而言,由于其基因组的高度变异性和杂合性,在病毒基因组中尚未发现保守序列设计出通用引物,用于环境病毒基因多样性解析[62]。但有研究发现,针对一些特定病毒家族,其基因组中编码某些结构或功能蛋白的氨基酸片段序列高度保守,可基于此序列设计出简并性引物,然后从环境中提取eDNA,直接进行PCR扩增目标基因,测序分析特定病毒家族遗传基因多样性及分布格局[63]。2014年Adriaenssens和Cowan[64]在一篇综述文章中归纳出多个病毒标记基因可用于解析环境特定病毒多样性及其生态学研究。需要说明的是这些标记基因多是针对海洋、湖泊等水体环境中细菌病毒基因多样性研究的,适用于土壤细菌病毒研究最常用的标记基因是g23,该基因是编码T4型噬菌体主要壳蛋白的结构基因。该基因最先是由Filée等[65]发现可用于研究海洋中T4型噬菌体多样性,随后Jia等[66]证明g23基因也可适用于研究稻田土壤中T4型噬菌体基因多样性,从而开启了g23基因在土壤病毒研究中应用的序幕[67-68]。针对该基因,中国科学院东北地理与农业生态研究所农田分子生态学科组从我国东北稻田、湿地和黑土农田中捕捉到数百条不同的T4型噬菌体g23基因,发现旱地黑土农田T4型噬菌体分布与其在海洋和湖泊水体环境中明显不同,而与稻田环境相近,从黑土农田获得的基因序列中建立了多个新的T4型噬菌体g23基因群[69-70]。发现即使在相同稻田环境,中国东北稻田T4型g23基因群集(Assemblage)与日本稻田不同,由此得出T4型噬菌体分布受到地理分隔和生态过程的双重调控的观点[70-71]。此外,我们还对g20基因(编码蓝藻肌尾噬菌体壳组装蛋白)[72-73]、DNApol基因(编码蓝藻短尾噬菌体DNA聚合酶基因)[74-75]、辅助代谢基因psbA(编码蓝藻噬菌体光合蛋白基因)[76]和phoH(编码噬菌体磷酸盐调节基因)[77-78]在东北稻田和湿地中基因组成及多样性进行了研究。揭示出这些标记基因表征的噬菌体群落结构组成在稻田和湿地环境中与海洋环境完全不同,并且在湿地与稻田间也存在差异,建立了多个独特的稻田和湿地噬菌体类群。由此可见,一些用于研究海洋环境噬菌体多样性的标记基因也适合用于陆地生态环境,说明细菌病毒有着共同的起源祖先;而基于分子标记基因群集表征的特定噬菌体群落结构在海洋、湿地、稻田,乃至旱地土壤中的不同,也从一个侧面显示出细菌病毒从海洋到陆地生态系统的演替过程。
2.2 RAPD-PCR解析病毒多样性
由于尚未发现通用引物适应于病毒多样性研究,如何解析环境特别是土壤中病毒多样性是一个挑战性课题。随机扩增多态性DNA标记(Random Amplified Polymorphic DNA ,RAPD)是采用随机的10个碱基核苷酸序列同时作为正向和反向引物进行PCR扩增,是对未知序列基因组进行多态性分析的技术[79]。Winget和Wommack[80]首次将该技术运用到美国切萨皮克湾水体病毒群落研究,他们根据扩增产物凝胶电泳图谱中条带的位置(长度)、亮度(丰度)分布情况,分析出不同样本间病毒群落结构及其相似度,然后对条带进行切胶测序,分析病毒组成。这种分析方法与DGGE-PCR技术解析环境微生物群落结构流程基本类似。利用该技术,Helton和Wommack[81]进一步分析了切萨皮克湾沉积物中病毒基因多样性,发现沉积物病毒结果存在明显的时空分变化;Srinivasiah等[82]揭示出南极不同地点土壤病毒群落结构存在明显差异。中国科学院沈阳应用生态研究所徐慧团队[83]采用该技术并结合高通量测序技术,对长期灌溉和施用尿素的稻田土壤中病毒和细菌群落丰度及多样性进行动态解析,发现添加尿素延迟了病毒丰度峰值的出现,说明病毒对氮肥添加非常敏感。需要注意的是,运用该技术的前提条件是要确保提取到的DNA没有寄主细胞遗传物质的干扰,然后才可进行PCR扩增。此外,也要考虑到单个随机引物扩增的偏好性,会导致在对病毒群落结构解析时存在偏差。故此,应用该技术时,建议采用多个随机引物进行扩增,然后将不同引物扩增得到的凝胶电泳条带图谱整合为一个数据集,再进行病毒群落结构解析。
2.3 宏基因组学解析病毒多样性
虽然RAPD-PCR技术在一定程度上能够对不同样本病毒群落结构进行解析,但该技术尚不能对病毒成员进行定性描述,还需后期大量的条带切胶测序工作。随着基因测序技术的飞速发展,高通量测序技术已成为研究环境微生物组的主流技术。其中宏基因解析技术目前成为不依赖于特定引物扩增的环境病毒组研究的热点领域[84-87]。与RAPD-PCR研究相同,环境病毒宏基因组学解析的前提条件是去除细胞生物遗传基因在病毒遗传物质中的污染。为此,研究者们在病毒基因提取、纯化等方法学研究上开展了许多工作,包括利用不同孔径微孔滤膜的切向过滤法(TFF)、核酸酶消化处理去除病毒浓缩液中细胞生物遗传物质、10%聚乙二醇(PEG)溶液沉淀浓缩病毒颗粒、浓度为2.9 mg·L-1的FeCl3溶液作为病毒絮凝剂的絮凝-过滤-再悬浮方法(FFR)、20%蔗糖垫超离心纯化法或不同密度CsCl溶液超离心纯化法等[85,88-90]。需要指出的是,这些方法对环境病毒的分离过程的第一步均是采用将提取到的病毒溶液通过0.2 μm的微孔滤膜而富集,这一步骤无疑会将一些尺寸较大的病毒,如巨大病毒等排出在外[91]。2009年Nature Protocols上发表一篇文章,详细介绍了针对水体样品宏病毒基因组学研究的方法[90],该文研究策略可推广应用到土壤病毒宏基因组解析,不同之处在于土壤有前处理,也就是从土壤中高效地分离提取出病毒溶液的过程。在开展环境宏病毒基因组学研究另一个需要注意的事项是提取到的病毒遗传物质量的多少。虽然环境病毒数量明显高于寄主生物,但由于提取效率低和病毒基因组小的原因,从常规的样本用量(如水样20 L,土样10~50 g)中提取到的病毒遗传物质量很少,不能满足后续宏基因组研究的需要,需要经过体外扩增技术以增加病毒遗传物质的浓度,达到测序的要求。目前常用的方法有两种:一是多重置换扩增(MDA)技术,即采用φ 29 DNA聚合酶进行扩增技术[87,90];二是连接扩增(LASL)技术[92]。需要引起注意的是两种扩增技术在扩增产物上存在偏好性,采用MDA倾向于扩增出ssDNA病毒,而采用LASL得到的病毒数据全部来自于dsDNA病毒[93]。其他研究土壤病毒宏基因组结果也证实MDA法倾向于获得大量的ssDNA病毒[94-95]。这样的结果与土壤中实际情况不符合,因为土壤病毒主要以噬菌体为主,且以dsDNA的有尾噬菌体占有较高的比例。为了避免体外扩增产生的偏好性,有研究者采用加大土壤样品用量,甚至到几千克,然后从土壤中大量富集病毒颗粒,提取病毒遗传物质,直接进行宏基因组测序的方法[96-97]。需要指出的是,上述研究方法主要是针对DNA病毒,对于环境RNA病毒宏基因组研究则需要采取其他的技术,通常采用的是RP-SISPA(Random Priming-mediated Sequence-Independent Single Primer Amplification)基因扩增,然后再进行高通量测序[98-99]。图2汇总了基于DNA水平对土壤病毒研究的主要方法及流程图。

图2 土壤病毒研究的主要方法及流程图Fig. 2 Main methods used in researches on soil viruses and their flowcharts
制约环境病毒宏基因组研究的另一个瓶颈问题是对基因数据解析的困惑。与细菌等微生物数据库相比,病毒基因数据库非常匮乏,从而导致绝大多数环境病毒宏基因组序列缺少参比对象,基因来源不清,被划分为未知基因,即使在剩下已比对上的基因中,也仅有非常小的一部分被确定为病毒,大部分基因与来源于细胞生物基因,尤其是细菌具有较高的同源性[84,87,100]。例如中国科学院生态环境研究中心贺纪正团队对海滩和稻田土壤的病毒宏基因组分析发现,仅9.4%~40.5%的序列可以被注释为已知的微生物信息,而在这些已知信息中被注释为病毒的占到82%~97%[101]。Trubl等[86]对瑞典一个冻融地带泥炭和沼泽土壤病毒宏基因组分析发现,尽管采用了严格的去除寄主遗传物质的步骤,但仅19%的序列被鉴定为病毒。Williamson等[37]总结国际上8个土壤病毒宏基因组数据库发现,有54.5%~97.3%的序列为未知来源和功能的序列。同样,最近中国科学院生态环境研究中心韩丽丽团队[102]对玉米根际与非根际土壤病毒宏基因组分析发现,仅0.26%~1.54%的序列被确认为病毒。大量源自病毒宏基因中的序列为未知基因,所以病毒也被喻为生物“暗物质”[57]。由此可见,高比例的环境病毒宏基因序列与已知的病毒数据不匹配,一定程度上妨碍了对病毒群落结构及生态功能的深入解析,这也反映出对环境病毒分离、纯化和测序的重要性。只有基于病毒基因数据库不断扩大、完善基础上,对环境病毒宏基因组解析才能够更全面、更深入。目前国外学者相继开发出多款数据分析软件,如VIROME、VirSorter、Metavir和VirusSeeker等,用于环境病毒宏基因组数据解析[103-106]。
3 土壤病毒的生态功能
虽然土壤病毒数量巨大且广泛存在,但我们对土壤病毒生态功能的认知尚处于初级阶段,主要的功能理解多源自病毒基础知识和海洋生态系统的相关研究结果,尚缺乏直接源自土壤中的实验佐证。2018年德国哥廷根大学的Kuzyakov和Mason-Jones[15]撰文从5个角度对土壤病毒的生态功能给予概念性的描述:1)病毒路径(Viral shunt):即土壤微生物被病毒感染裂解死亡,导致微生物中的碳氮磷硫等营养元素释放出来被其他微生物和植物利用的过程。在海洋中由病毒驱动的碳循环量占海洋生态系统碳循环总量的6%~26%[33],在土壤环境中,营养元素循环在多大强度上受到病毒驱动还鲜见报道;2)促进土壤微生物快速周转,保持活力状态(Forever young):即由于土壤病毒的感染,导致土壤细菌快速更替,从而使存活的细菌始终处于年轻状态,具有旺盛的代谢活力;3)病毒调节“胞外酶代谢”(Viral regulatory gate of EXOMET)。胞外酶代谢是指微生物细胞体内酶释放到环境中,起到分解有机物质的作用。这个概念是2013年Maire等[107]首次提出的,他们发现经过γ-射线处理几周的土壤已经没有微生物存活,但这些土壤培养后还释放出相当于未照射土壤处理的20%~60% CO2排放量。从这个现象中,我们不难理解土壤病毒感染微生物后,导致微生物裂解、胞内酶释放到土壤中,间接地促进了土壤有机物分解转化;4)通过微生物死体稳定性转化,促进土壤固碳作用(C sequestration by microbial necromass stabilization)。土壤有机质是土壤碳库稳定存在形式,Liang等[108]分析发现50%以上的土壤有机质来源于微生物死亡细胞,即通过微生物碳泵的续埋作用(Entombing effect)促进土壤固碳[109]。病毒导致微生物死亡,形成了大量的残体碎片,可能促进了土壤有机质形成,起到固碳作用。需要说明的是,微生物细胞被病毒裂解后形成的死亡残体与细胞受到非生物因素致死形成的残体,哪种途径更有利于促进土壤有机质形成尚无答案;5)引起土壤微域尺度C、N和P化学计量学分异(Microscale divergence of C/N/P stoichiometry)。这种作用主要是基于寄主细胞和病毒之间C、N和P化学计量学比值的差异,细菌病毒较细菌富含N和P,尤其是P元素。病毒感染细菌细胞裂解后,细菌细胞中有大约10%的碳、15%的氮和33%的磷营养转变成病毒颗粒成分,不能被植物和其他生物利用,从而影响生态系统的养分循环。这种由于微生物和病毒之间元素化学计量差异而引起的磷营养限制仅在海洋环境中有报道[48],在土壤环境中,由于病毒生物量很小,以及土壤中养分含量相对较高的缘故,笔者推测病毒通过这种方式影响土壤养分循环的作用可能微乎其微。
上述土壤病毒功能主要是基于病毒基础知识认知推测得来的。从病毒生态学角度来分析,土壤病毒还有以下几方面功能:1)土壤病毒起到由上而下(Top-down)调控微生物群落结构的作用,从而间接地影响到土壤生态功能[110-111]。如向土壤中接种烈性根瘤菌噬菌体能够降低根瘤菌数量,从而降低根瘤量[112];向土壤中接种青枯病菌噬菌体,通过噬菌体疗法能够降低作物青枯病的危害[113]等;2)土壤病毒还是基因水平移动的载体,病毒与寄主之间不断地进行基因交流或突变,使得病毒和微生物获得新的性状和功能,共进化推动了地球生物群落不断演替[46];3)土壤病毒基因组携带辅助代谢基因(Auxiliary metabolic genes,AMGs),协助寄主微生物驱动生物地球化学循环。如最近对红树林土壤和农田土壤研究均发现病毒宏基因组中含有多种水解多糖类物质的CAZymes基因,推测这些基因在促进土壤碳循环中起到重要作用[102,114];4)土壤病毒可通过介导抗性基因的水平转移及调节宿主菌代谢通路来协助宿主抵抗低营养、辐射和污染等多种生态胁迫,对维持受胁迫生态环境中的微生物群落结构及功能稳定具有重要意义[115-116]。但目前关于低营养胁迫下的这一功能进行了较多研究,而针对污染环境(如农药污染、石油污染和重金属污染等)中,噬菌体协助宿主适应环境的相关研究还很少。图3概况了以土壤病毒为中心,病毒起到的直接或间接生态功能。
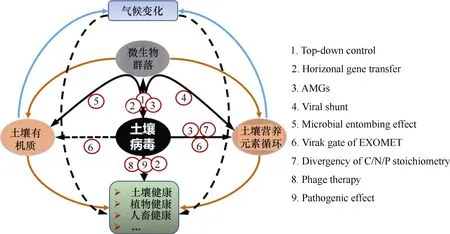
图3 土壤病毒主要生态功能示意图Fig. 3 Diagram of the main ecological functions of the viruses in soil
4 展 望
土壤中蕴藏着巨大的生物多样性,仅以原核生物为例,其在土壤中的多样性较其他生态系统的总和高出3个数量级[117-118],鉴于土壤微生物多样性高和巨大的生态功能,世界许多国家相继实施了一系列重要的土壤微生物组研究计划,中国科学院也于2014年启动了“土壤-微生物系统功能及其调控”战略先导专项,取得了一系列重要发现和成果[119]。这些研究主要涉及到具有细胞结构的微生物,很少涉及土壤病毒。进入21世纪,海洋领域病毒生态学研究发展迅速,科学家们发现病毒尤其是噬菌体在海洋生态系统物质循环、能量流动,以及维持海洋生物多样性和生物进化等方面起到重要的作用[31,52,120-121]。与海洋研究相比,针对土壤病毒研究却进展缓慢[11,37]。鉴于土壤病毒存在状态,以及土壤种类多样性和土壤环境的高度异质性,土壤中病毒组成较海洋等水体环境更复杂、多样性更高,研究的挑战性也会更大[12]。目前世界各国科学家已意识到了土壤病毒的重要作用和研究的紧迫性,相继发表了一些综述性文章报道土壤病毒研究进展,呼吁学术界对土壤病毒研究的重视[11-13,15,37,122]。本文梳理了一些土壤病毒研究的最新进展,从这些研究结果看,我们对土壤病毒的认知还很有限,尤其是对土壤病毒生态功能展示方面还缺乏直接的证据,缺乏将土壤病毒群落与寄主群落演替耦合起来的研究策略。基于此,认为未来对土壤病毒研究应在以下方面给予加强。
4.1 重视对土壤RNA病毒的研究
目前多数研究结果是针对土壤DNA病毒,较少涉及到土壤RNA病毒,而许多侵染真核生物的病毒是RNA病毒,尤其是导致人、畜和家禽流行性疾病的病毒,以及绝大多数的引起植物病害的病毒都是RNA病毒,如引起新冠肺炎病毒、各种禽流感病毒、烟草花叶病毒和马铃薯Y病毒等。有关这些病原性的RNA病毒在土壤中生存状态、迁移,以及活性受那些土壤因素影响,是否具有感染能力等问题还缺少系统性的研究。
4.2 关注对土壤溶原性噬菌体的研究
现阶段对土壤病毒研究多采用基于微孔膜过滤富集病毒颗粒的方法,这种方式无疑将大部分溶原性噬菌体屏蔽在外,而土壤中溶原性噬菌体是土壤病毒主要存在形式,缺乏对溶原性噬菌体的研究,也限制了对土壤整体病毒多样性及生态功能的认知。
4.3 关注土壤病毒资源的开发,提高土壤病毒研究的市场经济价值
土壤病毒不仅是最大的基因宝库,许多病毒资源也具有非常大的应用价值,如从土壤环境中筛选出能够高效裂解动植物致病细菌的噬菌体资源,应用噬菌体疗法防控动植物病害是一个非常有前景的研究领域[123-124]。此外,病毒还是天然的生物纳米材料,其不但拥有纳米材料的优越性能,还具备可快速复制的优势,同时其遗传物质较小,易于被基因工程技术改造,因此被越来越广泛地应用到纳米材料生成技术的各个方向,包括显示屏和电池等[125-126]。
4.4 注重对微生物病毒纯培养株的分离及基因组解析研究
目前针对病毒纯分离株全基因组的解析量远小于具有细胞结构的微生物,导致病毒全基因组数据库量小,大量土壤病毒宏基因组数据无法判断来源,妨碍了后续解析工作。因此在今后研究过程中,在采用分子生物学技术研究土壤病毒生态学问题的同时,也要关注微生物病毒纯培养株全基因组分析工作,尤其是对古菌病毒和真菌病毒株的研究。这些基础性的工作是扩大环境病毒数据库的前提,是开发新分析软件和建立新分析平台的保障,也是开启土壤病毒基因宝库的钥匙。
4.5 加强土壤病毒与土壤微生物的耦合研究
除部分土壤病毒能够直接引起动植物病害和人类疾病外,土壤病毒主要是通过调控寄主微生物群落结构组成、影响群落演替而间接地体现出病毒的生态功能。目前国际上对土壤病毒的研究还局限于盘点其多样性和数量丰度等层面,对病毒生态功能的研究还很少。多数对土壤病毒的研究未能与土壤微生物群落变化联系起来,这无疑会妨碍我们对病毒生态功能的认知。故此,在今后的研究中,应加强对土壤病毒与土壤微生物的耦合研究,通过精巧的控制实验设计,研究揭示出病毒在构建健康土壤环境、调控根际微生态、促进植物生长,乃至影响全球气候变化等方面的重要作用。
