希特林蛋白缺陷病并自身免疫性溶血1例及文献复习
2020-11-28段金涛邓成俊李娟
段金涛 邓成俊 李娟
【摘要】 目的 对1例临床诊断为新生儿肝内胆汁淤积症(NICCD)合并自身免疫性溶血的患儿及家系进行SLC25A13基因突变分析, 以达到早确诊早治疗的目的。方法 结合患儿的临床特点及靶向捕获二代测序(NGS)检测结果总结患儿的遗传特点进行确诊。结果 患儿表现为皮肤、巩膜黄染等症状, 伴血乳酸、甲胎蛋白等明显升高, 临床诊断为NICCD。基因检测结果提示患儿SLC25A13基因5号外显子及5号内含子存在长片段纯合变异, 父母均为携带者。结论 NICCD合并自身免疫性溶血病患临床上较为罕见, 本文报告了该疾病致病基因新的突变形式, 为日后疾病的及时诊疗提供案例参考。
【关键词】 希特林蛋白缺陷病;SLC25A13;肝内胆汁淤积症
DOI:10.14163/j.cnki.11-5547/r.2020.29.077
【Abstract】 Objective SLC25A13 gene mutation analysis was performed on a child and family with a clinical diagnosis of neonatal intrahepatic cholestasis caused by citrin deficiency (NICCD) combined with autoimmune hemolysis to achieve the purpose of early diagnosis and early treatment. Methods Combined with the clinical characteristics of the children and the results of Next generation sequencing (NGS) detection, the genetic characteristics of the patients were summarized for diagnosis. Results The children presented with symptoms such as skin and sclera yellow staining, accompanied by a significant increase in blood lactic acid and alpha-fetoprotein, and was clinically diagnosed as NICCD. The results of genetic testing indicated that there were long homozygous mutations in exon 5 and intron 5 of SLC25A13 gene, and both parents were carriers. Conclusion NICCD combined with autoimmune hemolytic disease is clinically rare. This article reports a new mutation form of the disease-causing gene, which provides a case reference for timely diagnosis and treatment of the disease in the future.
【Key words】 Citrin deficiency; SLC25A13; Neonatal intrahepatic cholestasis caused by citrin deficiency
希特林蛋白缺陷病(citrin deficiency, CD)是一種由于SLC25A13基因缺陷导致希特林蛋白表达异常的功能缺陷性疾病, 为常染色体单基因隐性遗传疾病[1]。致病基因位于染色体7q21.3, 含18个外显子和17个内含子, 由675个氨基酸组成, 全长约200 kb, 主要在肝细胞内表达。目前已报道CD具有3种年龄依赖性临床表型:希特林缺陷导致的新生儿肝内胆汁淤积症(neonatal intrahepatic cholestasis caused by citrin deficiency, NICCD)、成人发病瓜氨酸血Ⅱ型(adult-onset type Ⅱ citrullinemia, CTLN2)、希特林缺陷导致的生长发育落后和血脂异常(failure to thrive anddyslipidemia caused by citrin eficiency, FTTDCD)[2]。其中NICCD是目前CD儿科主要的临床表型, 本研究对1例合并自身免疫性溶血的NICCD患儿进行分析及文献复习, 报告如下。
1 临床资料
患儿女, 2月6天, 彝族, 因“皮肤黄染1月余, 加重4 d”入院。患儿生后1周出现皮肤、巩膜黄染, 当地医院先后予蓝光箱等治疗效果不佳, 4 d前家长发现患儿黄疸较前加重就诊。近1周无发热, 无嗜睡、抽搐, 无咳嗽、呕吐, 否认白陶土样大便。查体:皮肤、巩膜黄染, 无皮疹及瘀斑、淤点, 腹膨隆, 肝右肋下3 cm, 剑突下1.5 cm可及, 脾未及肿大。辅助检查:血型AB型, RhD阳性。血常规检测:白细胞计数 13.97×109/L, 红细胞计数3.12×1012/L, 血红蛋白 77.00 g/L, 网织红细胞百分比5.36%, 红细胞形态:成熟红细胞轻度大小不等, 中心浅染区扩大。Coombs试验:直接抗人球蛋白试验:+;间接抗人球蛋白试验:+;抗IgG:阳性;抗C3:阴性。肝功能:谷丙转氨酶 67 U/L, 谷草转氨酶 122 U/L, 总胆红素 209.3 μmol/L, 间接胆红素 74.5 μmol/L, 直接胆红素 134.8 μmol/L, 总胆汁酸 166.7 μmol/L, 总蛋白49.9 g/L, 白蛋白27.9 g/L。
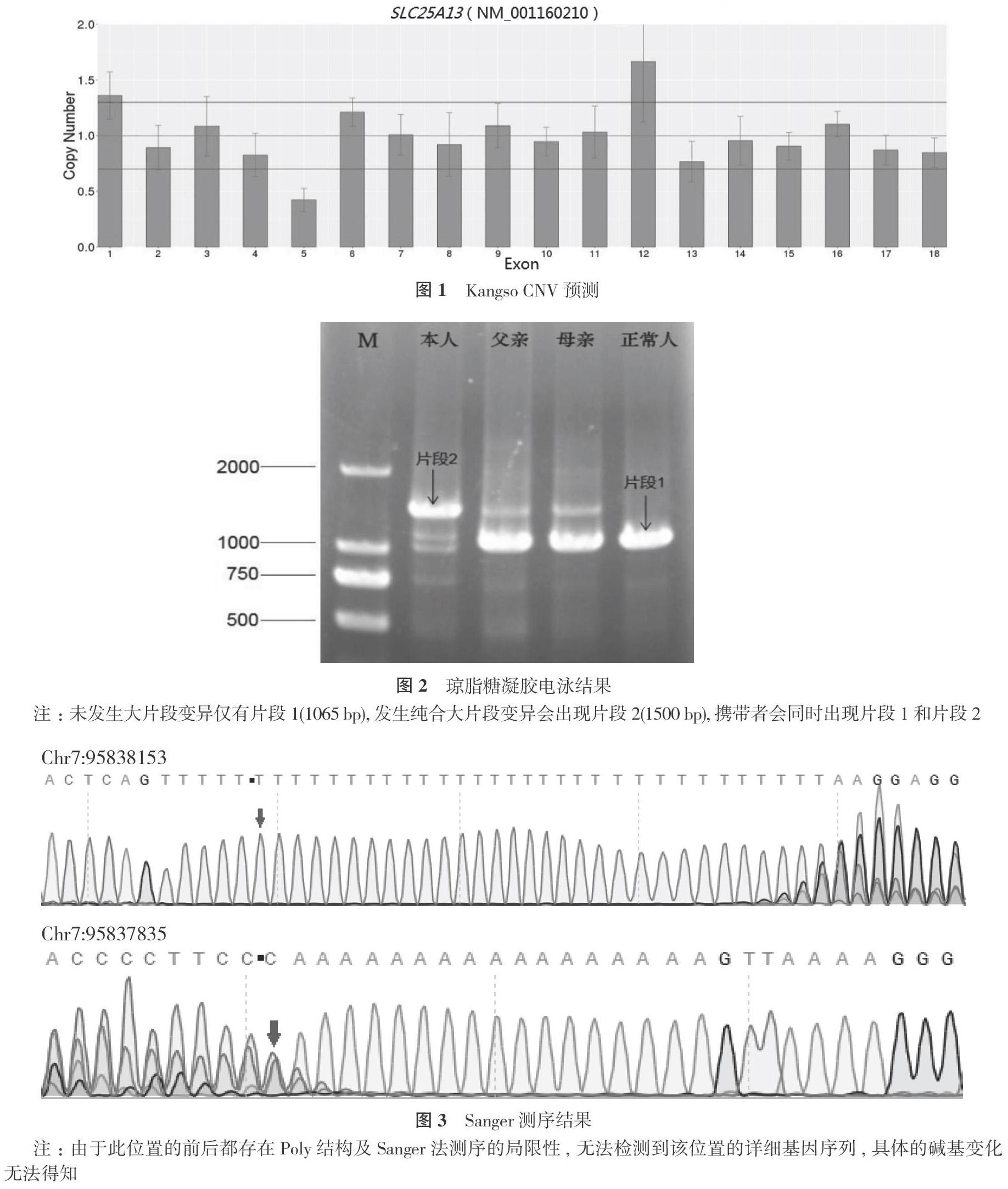
血氣分析:pH 7.445, 碱剩余(BE) -8.1 mmol/L, 乳酸浓度3.2 mmol/L。甲胎蛋白 60500 ng/ml。空腹血糖2.2 mmol/L。血串联质谱检测为瓜氨酸102.64 mmol/L、甲硫氨酸119.66 mmol/L、酪氨酸 66.04 mmol/L、瓜氨酸/苯丙氨酸比值2.96、甲硫氨酸/苯丙氨酸比值3.45, 谷氨酸/瓜氨酸比值0.91。结合患儿血乳酸、甲胎蛋白等明显升高, 有低蛋白、低血糖等表现, 临床诊断为NICCD, 予无乳糖和富含中链甘油三酯的配方奶粉喂养2周, 复查血串联质谱检测瓜氨酸、甲硫氨酸、酪氨酸等均降至正常范围, 3个月后检查肝功能、胆红素基本正常。临床诊治过程中, 经昆明市儿童医院医学伦理委员会审核批准, 遵循家属意见并签署知情同意书后, 采集患儿及父母外周血2 ml, 送北京康旭医学检验所进行全外显子检测, 重点关注希特林蛋白缺陷病, 并根据拷贝数变异(CNV)预测结果, 进一步完善SLC25A13基因检测, 结果提示SLC25A13基因Exon5的全部或部分可能存在变异(见图1), 后采用Sanger法测序及琼脂糖凝胶电泳, 证明患儿SLC25A13基因5号外显子及5号内含子(chr7:95837835-95838153位置)存在长片段纯合插入变异(见图2, 图3), 其父母为长片段变异的携带者(杂合变异, 见图2)。
2 讨论
Citrin蛋白是一种由SLC25A13基因编码的线粒体内钙结合蛋白, 其功能是将线粒体内的天冬氨酸转运至胞质内, 参与尿素、蛋白及核苷酸的合成, 将还原型烟酰胺腺嘌呤二核苷酸(NADH)转变为烟酰胺腺嘌呤二核苷酸(NAD+), 而NADH/NAD+比值是维持部分细胞生化代谢途径的重要因素[3, 4]。Citrin功能缺陷可导致细胞质内NADH堆积, 蛋白质与核酸合成受到抑制, 引起一系列生化代谢紊乱。
NICCD临床相对少见, 其发病率为1/34000~1/17000, 男女比例无明显差异, 多数新生儿期起病[3-5], 主要的临床表现为黄疸、肝大、脂肪肝、生长发育迟缓等;生化检查可见肝酶异常、胆汁淤积、低蛋白血症及低血糖, 部分患儿存在凝血功能异常, 血氨、甲胎蛋白、乳酸升高或血脂异常[3, 6]。目前缺乏公认的临床或生化诊断标准, 基因分析存在2个SLC25A13等位基因纯合或复合杂合突变可确诊。
本例患儿以皮肤黄染加重就诊, 入院经血液、生化检查发现存在“自身免疫性溶血”, 对症治疗后溶血停止, 但患儿同时伴有谷丙转氨酶、谷草转氨酶、直接胆红素、总胆汁酸、甲胎蛋白、血乳酸等升高, 白蛋白和空腹血糖降低, 自身免疫性溶血不能完全解释患儿临床表现, 结合血串联质谱检测结果及予无乳糖和富含中链甘油三酯的配方奶喂养后病情明显好转, 临床诊断NICCD, 进一步完善SLC25A13基因检测, 发现5号外显子及5号内含子存在长片段纯合插入变异, 其父母为变异携带者, 符合常染色体隐性遗传方式, 确诊患者为NICCD合并自身免疫性溶血, 这在临床比较罕见, 既往相关文献未见报道[4, 5, 7, 8]。
目前, 全世界已发现100余种SLC25A13基因突变, 我国以c.851-854del4(58.1%)、c.1638-1660dup23(8.85%)、IVS6+5G>A(8.41%)和IVSl6ins3kb(7.52%) 4种突变最为常见, 占80%以上[9, 10], 但随着新的检测方法在临床的应用, 新的突变位点不断报道[11], 同时, 部分确诊的患儿也发现存在一些之前未被发现的特殊临床表现[12, 13]。本研究检出的突变位点既往在人类基因突变数据库(HGMD)、EnSembl数据库、千人基因组数据库中均未见报道, 对于完善我国SLC25A13基因突变数据库具有重要意义, 但目前尚不能肯定新突变与临床表现的相关性, 还需通过动物模型或蛋白功能表达等研究进一步证实。同时, 在临床工作中, 当临床医师在遇到无法用一种疾病解释病情时, 需要医生仔细结合病史、查体及辅助检查, 及时进一步完善相关检查, 避免漏诊、误诊, 也能够为患儿临床早期遗传咨询及明确病因提供参考。
参考文献
[1] Zhang ZH, Lin WX, Zheng QQ, et al. Molecular diagnosis of citrin deficiency in an infant with intrahepatic cholestasis: identification of a 21. 7kb gross deletion that completely silences the transcriptional and translational expression of the affected SLC25A13 allele. Oncotarget, 2017, 8(50):87182-87193.
[2] Okano Y, Ohura T, Sakamoto O, et al. Current treatment for citrin deficiency during NICCD and adaptation/compensation stages: Strategy to prevent CTLN2. Mol Genet Metab, 2019, 127(3):175-183.
[3] Lu CT, Shi QP, Li ZJ, et al. Blood glucose and insulin and correlation of SLC25A13 mutations with biochemical changes in NICCD patients. Exp Biol Med (Maywood), 2017, 242(12):1271-1278.
[4] Ohura T, Kobayashi K, Tazawa Y, et al. Clinical pictures of 75 patients with neonatal intrahepatic cholestasis caused by citrin deficiency (NICCD). J Inherit Metab Dis, 2007, 30(2):139-144.
[5] Oh SH, Lee BH, Kim GH, et al. Biochemical and molecular characteristics of citrin deficiency in Korean children. J Hum Genet, 2017, 62(2):305-307.
[6] Saheki T, Inoue K, Tushima A, et al. Citrin deficiency and current treatment concepts. Molecular Genetics & Metabolism, 2010, 100(supp-S):S59-S64.
[7] 彭曉康, 刘希, 刘攀, 等. Citrin缺陷导致的新生儿肝内胆汁淤积症32例临床特点分析. 中华实用儿科临床杂志, 2019, 34(5):377-380.
[8] 白欣立, 张亚男, 杨亭亭, 等. Citrin缺陷导致新生儿肝内胆汁淤积症患儿临床特点及SLC25A13基因分析. 中国妇幼保健, 2017, 32(19):4748-4751.
[9] Song YZ, Zhang ZH, Lin WX, et al. SLC25A13 gene analysis in citrin deficiency: sixteen novel mutations in East Asian patients, and the mutation distribution in a large pediatric cohort in China. PloS one, 2013, 8(9):e74544.
[10] Zhang ZH, Lin WX, Deng M, et al. Clinical, molecular and functional investigation on an infant with neonatal intrahepatic cholestasis caused by citrin deficiency (NICCD). PloS one, 2014, 9(2):e89267.
[11] Lin WX, Zeng HS, Zhang ZH, et al. Molecular diagnosis of pediatric patients with citrin deficiency in China: SLC25A13 mutation spectrum and the geographic distribution. Sci Rep, 2016(6):29732.
[12] Lipiński P, Jurkiewicz D, Ciara E, et al. Neonatal cholestasis due to citrin deficiency: diagnostic pitfalls. Acta biochimica Polonica, 2020, 67(2):225-228.
[13] 张建玲, 舒赛男, 蔡在胜, 等. 以肝硬化腹腔积液为特征表现的Citrin蛋白缺陷所致新生儿肝内胆汁淤积症1例报告. 临床肝胆病杂志, 2019, 35(2):372-375.
[收稿日期:2020-06-24]
