基于一致性学习预测药物-靶标相互作用
2020-11-27
(湖南工业大学 计算机学院,湖南 株洲 412007)
1 研究背景
药物研发是现代社会中的一个重要课题[1]。通常情况下,新药研发从确定思路到投入市场大约需要花费12~15 a 的时间,成本约10 亿美元,且成功率低于10%[2]。据统计,美国食品药品监督管理局(food and drug administration,FDA)每年批准能作为新药的分子大约只有20 种[3]。诺贝尔奖获得者James Black 认为,从旧药研发新药可能是研发新药的有效途径[4]。
药物重定位又称“老药新用”,是一种比较好的研发新药方法,可节约研发时间,降低研发成本,减少研发失败带来的风险,因而受到越来越多的关注。药物重定位是指发现现有药物的新用途,其生物学基础是一药多靶和一靶多药[5]。基于计算方法对药物-靶标相互作用进行预测,以发现某个药物的多个潜在靶标或某个靶标的多个药物,从而实现对现有药物的重定位,可以显著减少后期过程(如临床试验)的负荷,节省大量资源和时间。
药物-靶标相互作用预测方法可以分为基于网络的方法和基于机器学习的方法。其中,基于网络的方法把药物-靶标相互作用矩阵看成是一个网络,基于网络模型预测其潜在关联。Y.Yamanishi 等[6]把药物和靶标整合成统一的“药理空间”,提出一种二分图学习模型,用于推测靶标与药物之间的相互作用;M.Campillos 等[7]在“不相关药物的相似脱靶效应可导致药物的相似副作用”这一理论基础上,根据一体化医学语言系统(unified medical language system,UMLS)术语对药物的副作用进行了分类,提出衡量药物副作用相似性的方法,预测共享相同靶标的药物;K.Bleakley 等[8]基于药物的化学结构预测药物的药理相似性,然后利用局部二分图模型预测未知的药物-靶标相互作用;M.Gönen[9]使用全概率模型进行预测,把药物和靶标投影进统一空间,在此基础上整合贝叶斯模型,得到了KBMF2K(Kernelized Bayesian matrix factorization),再利用协方差计算中的变分近似进行推断。Cheng F.X.等[10]提出了3 种基于网络的推断方法,即基于靶标相似性的推断 方 法(target-based similarity inferrence,TBSI)、基于药物相似性的推断方法(drug-based similarity inferrence,DBSI)和基于网络的推断方法(networkbased inferrence,NBI),以预测可能的药物-靶标相互作用。
基于机器学习的方法提出机器学习相关模型,在确定模型性能后对药物与靶标之间潜在的关联进行预测。Y.Yamanishi等[11]基于药物的理化性质和药物-靶标相互作用网络的拓扑性质,提出了基于支持向量机的方法(supervised bipartite graph ingerrence,SBGI)挖掘可能的药物-靶标相互作用。S.Mizutani等[12]基于药物-靶标相互作用谱和副作用谱,定义稀疏典型相关分析进行预测。L.Jacob 等[13]利用核函数和支持向量机(support vector machine,SVM),研究了药物-靶标的相互作用。Xia Z.等[14]提出了一种基于流形正则化的半监督学习模型,以挖掘药物与靶标之间的潜在关联。T.Van Laarhoven 等[15]首先使用药物核函数和靶标核函数对相互作用数据进行稀疏化,然后定义药物相互作用谱和靶标相互作用谱,根据这些相互作用谱向量定义高斯核函数,并把高斯相互作用谱核与药物核和靶标核结合起来,利用正则化最小二乘法寻找能作用于已知靶标的新药。Chen H.L.等[16]根据与靶标的相关性对药物进行排序打分,提出采用网络一致性方法(network consistency-based prediction method,NetCBP), 推测药物-靶标的相互作用。T.VAN Laarhoven 等[17]首先定义了药物和靶标的相似性核,然后在正则化最小二乘法的基础上提出带权最近邻方法(weighted nearest neighbor-gaussian interaction profile,WNNGIP)。M.A.Heiskanen 等[18]对目前的预测方法进行了评估,发现基于排序的打分方法效果较好。Wang L.等[19]利用深度学习中的堆叠自动编码器,充分提取药物分子的化学子结构和靶标的蛋白序列特征,以识别药物-靶标相互作用半监督学习模型,有效识别了药物-靶标相互作用数据。机器学习方法有效预测了药物与靶标之间存在的关联,然而,由于缺少负样本数据,使得监督学习方法预测效果不够理想,而且大部分有监督预测方法的时间复杂度较高[13];同时,这些方法都严重依赖于药物的化学结构相似性和靶标的序列相似性[19-24]。
为了缓解这个问题,本文基于局部全局一致性学习方法(learning with local and global consistency,LLGC)[25],综合考虑靶标蛋白的序列特征和药物-靶标相互作用网络的拓扑性质,提出药物-靶标相互作用挖掘方法的LLGC,通过分析比较,发现所提LLGC 方法获得了较好的预测性能。
2 数据和模型
2.1 数据
本文使用的数据由Y.Yamanishi 等[6]提供,可从http://web.kuicr.kyoto-u.ac.jp/supp/yoshi/drugtarget/下载,具体包括药物相似性矩阵、靶标蛋白相似性矩阵及药物-靶标相互作用矩阵。通过KEGG GENES数据库[26]提取靶标的序列信息,基于正则化打分函数以度量其序列相似性:对于两个靶标t1与t2,假设SW(t1,t2)表示其Smith-Waterman 分值,则其序列相似性可用以下公式计算:
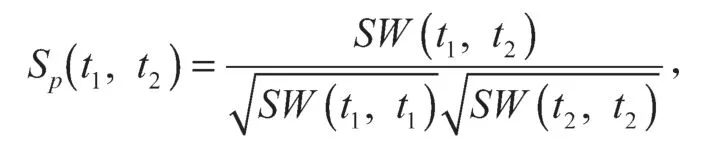
从而得到靶标之间的相似性矩阵Ssp。
Y.Yamanishi 等[6]分析DrugBank[27]、KEGG[28]和SuperTarget[29]等数据库发现,已知药物靶标的人类核受体、G 蛋白偶联受体(GPCRs)、离子通道和酶的数量分别是54,223,210,445 个,相对应的靶标蛋白分别为26,95,204,664 个,它们已知的相互作用数分别为90,635,1 476,2 926[6,10,13-18],详细情况如表1所示。在本实验中,使用Y.Yamanishi 等[6]提供的数据作为标准数据集,以评估模型的优劣。

表1 药物-靶标相互作用数据集Table 1 Drug-target interaction data set
2.2 模型
给定n个靶标和m个药物,若靶标i靶向药物j,则yij=1,否则yij=0,用矩阵Y=[y1,y2,…,yn]T表示靶标靶向药物的情况,即药物-靶标相互作用网络。如果多个靶标蛋白与药物相互作用,则靶标相似性程度可以由其共享药物数量来度量。因此,可以基于药物-靶标相互作用网络中靶标共享药物数,来度量其相似性:

综合序列相似性和药物-靶标网络的拓扑结构相似性,可计算如下靶标相似性矩阵:

式(1)(2)中:Snp为网络拓扑结构相似性矩阵;Ssp为序列相似性矩阵;参数μ用于衡量两者间的重要程度,且
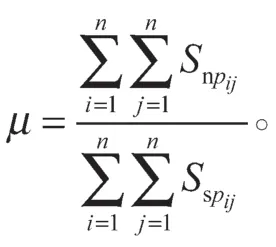
对Sp′进行归一化后得到:

用F∈{0,1}n×m表示预测到的药物-靶标相互作用矩阵。fij∈{0,1}表示经过学习后,模型预测到的靶标i与药物j的关联情况,若值为1,表示靶标i靶向药物j,否则靶标i与药物j没有关联。如果药物i与药物j非常相似,且靶标靶向药物i,则此靶标更有可能靶向药物j,F=[f1,f2,…,fn]T。
LLGC 学习认为:邻近的结点更有可能有相同的标签,这是数据的局部性问题;在流形结构或聚类中的结点更有可能具有相同的标签,这是数据的全局性问题。为探究新的药物-靶标相互作用,综合考虑靶标和药物数据的全局和局部特征,提出一致性药物-靶标相互作用预测算法LLGC,把已知的药物-靶标相互作用信息传播至其邻接点结点,从而获得新的药物-靶标相互作用。
算法1用迭代法挖掘药物-靶标相互作用
输入:靶标序列相似性矩阵,药物-靶标相互作用网络。
输出:新的药物-靶标相互作用矩阵F*。
Step 1 由公式(1)~(3)获得靶标相似性矩阵Sp,并令,以避免在标签学习过程中对结点进行了自增强处理。
Step 2 对靶标相似性矩阵进行正则化,得到拉普拉斯矩阵L=D-Sp,其中D是对角矩阵,第i个对角元素值为。
Step 3 对F(t+1)=αLF(t)+(1-α)Y迭代求解,直至收敛。α为介于0 和1 之间的参数。
Step 4 假设F*表示解集{F(t)}的权限,则F*为所求问题的解。
Step 5 输出预测的药物-靶标关联网络,预测药物-靶标相互作用。
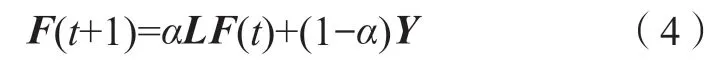
考虑算法1 中第t步的迭代模型的收敛性。模型第一部分接收来处邻接点的标签信息,第二部分用于保存初始标签信息。L=D-Sp,参数α∈(0,1),用于平衡来自邻接点的标签信息和初始标签信息之间的重要程度。
由公式(4)得

由0<α<1,L与矩阵相似,因而L的特征值在[-1,1]。从而

假设F(t)收敛于F*,对于药物-靶标相互作用预测问题,可以对其求极限,F*=limF(t),综合公式(5)~(7),可得:
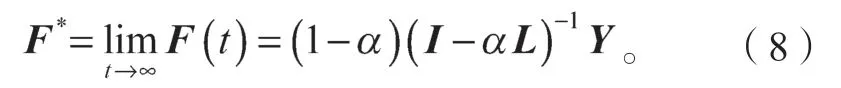
其中:I为n×n的单位矩阵;

不失一般性,公式(8)等价于下式:

根据公式(10),无需迭代,可直接计算出药物-靶标相互作用矩阵为

若用MT代替L,则可得到如下公式(11)的另一种形式:

结合M的定义,可得到另一药物-靶标相互作用矩阵:

综合公式(11)和(13),可得到如下发现药物-靶标相互作用的模型

从而在算法1 的基础上进行改进,得到算法2。
算法2基于LLGC 发现药物-靶标相互作用
输入:靶标序列相似性矩阵,药物-靶标相互作用网络。
输出:药物-靶标相互作用矩阵。
Step 1 根据公式(1)~(3),计算靶标相似性矩阵Sp,并令,以避免在标签学习过程中对结点进行了自增强处理。
Step 2 对靶标相似性矩阵进行正则化,得到拉普拉斯矩阵L=D-Sp,其中D是对角矩阵,第i个对角元素值为。
Step 3 根据Step 2 的计算结果求解和。
Step 4 根据公式(14)得到靶标的标签矩阵。
Step 5 输出药物-靶标相互作用矩阵。
3 实验结果和讨论
实验中,把药物-靶标相互作用数据以随机形式分成大小相等的5 份,依次选择1 份用作测试集,剩下的4 份用作训练集,实验重复20 次,取20 次实验的平均值作为最终结果。参数α取值0.9。
3.1 与其他方法分析比较
把预测方法LLGC 与SBGI、KBMF2K、NetCBP和WNN-GIP 进行分析比较,所得结果如表2所示。

表2 各方法的预测结果汇总表Table 2 Prediction performance of different DTI prediction methods
分析表2所示预测结果可以得知,LLGC 方法基于AUC(area under curve)的性能要好于SBGI 和KBMF2K 方法的。与SBGI 方法相比,4 个数据集中AUC 值分别提高了6.70%,17.49%,8.88%,1.60%;与KBMF2K 方法相比,4 个数据集中AUC 值分别提高了5.29%,1.75%,3.03%,0.36%;与WNN-GIP 和NetCBP 相比,除在核受体中AUC 值比WNN-GIP和NetCBP 稍低(低1.43%)之外,在AUC 上均好于其他4 种方法或与其他方法基本持平。
3.2 预测药物-靶标相互作用
课题组基于一致性学习,考虑靶标相似性和药物-靶标相互作用网络的拓扑性质,提出一种靶标相似性度量方法。在此基础上,考虑药物和靶标数据的全局一致性和局部一致性特征,提出LLGC 方法寻找药物-靶标相互作用。LLGC 方法将对每个药物与靶标之间的关系进行打分,并且根据得分对每一个药物-靶标对进行排序,从而预测药物-靶标的相互作用;然后,在DrugBank 数据库[27]、KEGG 数据库[28]和SuperTarget[29]数据库中,对预测到的药物-靶标相互作用进行分析验证。
表3~6 分别列出了核受体中、GPCRs 中、离子通道中和酶中预测到的分值最高的5 个药物-靶标相互作用对。

表3 核受体中预测到的排序前5 的药物-靶标相互作用对Table 3 Predicted top 5 drug-target interaction pairs in nuclear receptor
表3所示为核受体中预测到的排序前5 位的药物-靶标相互作用对,以靶标hsa2099 为例,预测到核受体中,靶标hsa2099 靶向药物D00182。在UniProt 数据库[30]中,靶标hsa2099 为核激素受体,调节真核细胞的基因表达,影响细胞的繁殖和目标组织的分化,调节涉及各种激酶的雌激素的信号传导。在SuperTarget、DrugBank 和KEGG 数据库中,检索到hsa2099 靶向药物D00066、D00067、D00105和D00554,其中D00066 是主要的孕激素类固醇,主要由黄体和胎盘分泌。孕激素作用于子宫、乳腺和大脑,有助于胚胎移植、妊娠维护和乳腺组织的发展。D00067 是哺乳动物的主要雌激素,雌激素酮是一种类固醇,主要产生于卵巢、胎盘和外围组织。D00105 是哺乳动物雌激素类固醇的一种形式,主要由卵巢和胎盘产生,可以合成各种同分异构体。D00554 也是一种具有较高的雌激素效能的口服药物。而D00182 可以合成孕激素,可用于治疗月经不调与子宫内膜异位等疾病。这说明靶标hsa2099 靶向药物D00182 有一定的根据。
运用LLGC 方法对核受体中的标准数据集进行预测,发现在90 个已知的药物-靶标相互作用 对中,有48 个药物-靶标相互作用对出现在预测集的前5%中,约占所有已知数据对的53.33%;69 个药物-靶标相互作用对在预测集的前10%中,约占所有已知数据对的76.67%;75 个药物-靶标相互作用对在预测集的前20%中,约占所有已知数据对的83.33%;所有90 个已知药物-靶标相互作用对在预测集的前50%中。

表4 GPCRs 中预测到的排序前5 的药物-靶标相互作用对Table 4 Predicted top 5 drug-target interaction pairs in GPCRs
表4为GPCRs 中预测到的排序前5 的药物-靶标相互作用对,同时在运用LLGC 方法对GPCRs 中的标准数据集进行预测过程中,发现在223 个已知药物-靶标相互作用对中,有93 个药物-靶标相互作用对出现在预测集的前5%中,约占所有已知数据对的41.70%;153 个药物-靶标相互作用对在预测集的前10%中,约占所有已知数据对的68.61%;172个药物-靶标相互作用对在预测集的前20%中,约占所有已知数据对的77.13%;203 个药物-靶标相互作用对在预测集中的前50%,约占所有已知数据对的91.03%。

表5 离子通道中预测到的排序前5 的药物-靶标相互作用对Table 5 Predicted top 5 drug-target interaction pairs in ion channel
表5为离子通道中预测到的排序前5 的药物-靶标相互作用对,同时在运用LLGC 方法对离子通道中的标准数据集进行预测过程中,发现在1 476 个已知药物-靶标相互作用对中,有514 个药物-靶标相互作用对出现在预测集的前5%中,约占所有已知数据对的34.82%;826 个药物-靶标相互作用对在预测集的前10% 中,约占所有已知数据对的55.96%;1 059 个药物-靶标相互作用对在预测集的前20%中,约占所有已知数据对的71.75%;1 344个药物-靶标相互作用对在预测集的前50%中,约占所有已知数据对的91.06%。
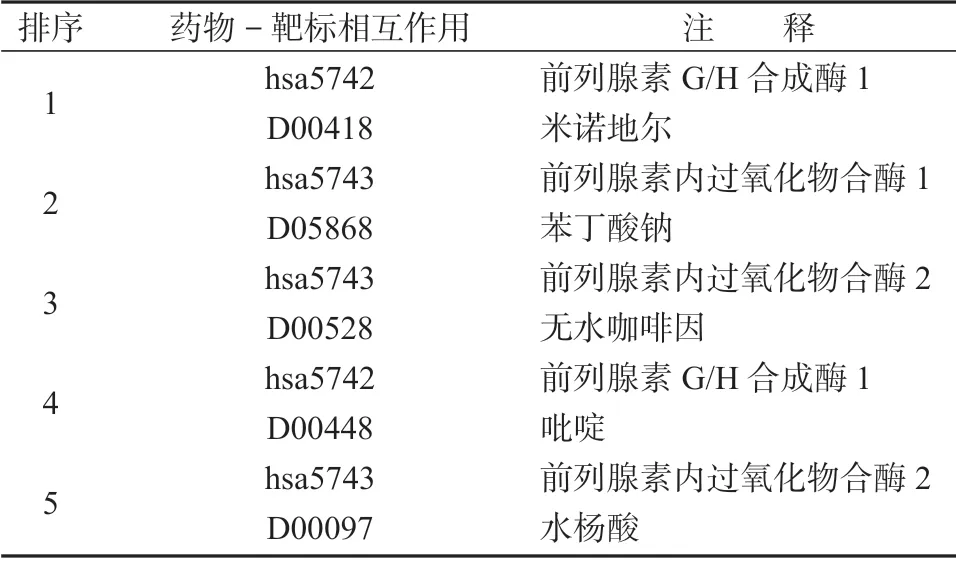
表6 酶中预测到的排序前5 的药物-靶标相互作用对Table 6 Predicted top 5 drug-target interaction pairs in enzyme
表6给出了酶中预测到的排序前5 的药物-靶标相互作用对,同时在运用LLGC 方法对酶中的标准数据集进行预测过程中,发现在2 926 个已知药物-靶标相互作用对中,有1 624 个药物-靶标相互作用对出现在预测集的前5%中,约占所有已知数据对的55.50%;有2 117 个药物-靶标相互作用对在预测集的前10%中,约占所有已知数据对的72.35%;2 489个药物-靶标相互作用对在预测集的前20%中,约占所有已知数据对的85.06%;2 926 个药物-靶标相互作用对在预测集的前50%中,即所有已知药物-靶标相互作用对都出现在预测集的前50%中。
4 结语
药物-靶标相互作用预测是现代药物研发中的一个重要课题。基于湿实验挖掘药物与靶标之间的相互作用耗时耗力,故需要开发相应的计算方法以发现这种潜在关联。因而本文基于一致性学习,融合靶标序列相似性和药物-靶标相互作用网络的拓扑性质,考虑数据的局部特性和全局特性,提出一致性药物-靶标相互作用预测方法LLGC,以预测来自人类核受体、GPCRs、离子通道和酶中的药物-靶标相互作用。与其他方法相比,LLGC 在AUC 上优于其他方法或与其他方法持平。同时,标准数据集中绝大部分已知的药物-靶标相互作用预测出现在预测集的前20%中,91%以上出现在预测集的前50%中。今后,课题组将结合深度学习相关模型和各种生物特征,以改进药物-靶标相互作用的预测性能。
