多孔BN选择性去除燃油中硫化合物的密度泛函理论研究
2020-10-27李巧灵吴晓宇王学伟谢智于晓飞杨晓婧黄阳李兰兰
李巧灵,吴晓宇,王学伟,谢智,于晓飞,杨晓婧,黄阳,李兰兰
(河北工业大学材料科学与工程学院,天津300130)
引 言
燃油中含硫化合物在高温燃烧时生成的硫氧化物(SOx)是形成酸雨和空气污染的主要因素之一,对环境和人类健康有极大的危害。目前,车用燃油的低硫和无硫化在世界范围内已是大势所趋,燃油的深度脱硫已经成为非常紧迫而亟需解决的世界性研究课题。
燃油中的有机硫化物主要以噻吩(thiophene,T)、苯并噻吩(benzothiophene,BT)以及其他噻吩衍生物形式存在[60%~70%(质量)],同时含有少量的硫醇、硫醚和二硫醚。目前,较为成熟的燃油脱硫技术有加氢脱硫(hydrodesulfurization,HDS)[1−2]、氧化脱硫[3−4]、生物脱硫[5]、萃取脱硫[6]和吸附脱硫[7−8]等。其中,HDS 是从化石燃油中去除硫化物最有效的方式,可高效去除硫醇、硫化物、二硫化物和一些噻吩类及其衍生物。但是,HDS 技术工作条件较苛刻,需要高温、高压、氢气环境和贵金属催化剂等,脱硫成本较高[9]。此外,HDS技术难以去除燃油中的BT、二苯并噻吩(dibenthiophene,DBT)及其衍生物等难降解的大分子硫化合物。而相对于传统加氢脱硫技术要求的高温、高压、氢环境以及贵金属催化剂等苛刻条件,吸附脱硫技术具有操作简单、快速、投资费用少、无污染、脱硫效果好等优点[10−11],是一项具有广阔发展空间及应用前景的新技术。
常见的吸附脱硫材料主要有活性炭[12]、金属有机骨架[13−14]、分子印迹聚合物[15]、氧化矿物材料等[16]。其中,活性炭具有大的比表面积、良好的孔结构、丰富的表面活性基团,且价格低廉,对有机硫和无机硫都有较好的脱除能力,是目前研究最为广泛的吸附剂[17]。但是,活性炭中C—C 非极性成键的特点,使其在吸附去除极性相对较强的噻吩类硫化物的同时,与油品中正十六烷、正辛烷、甲苯等可用非硫碳化物也有强相互作用,即选择性不高,易造成油品中高热量成分的损失。此外,活性炭在高温下容易氧化和晶化,导致吸附性能下降,限制了其高温或极端条件下的应用以及吸附后的再生。其他的脱硫吸附材料,由于材料本身的特点,也普遍存在选择性和再生性等方面的不足,无法有效去除噻吩类硫化物。因此,开发高选择性吸附脱硫材料,在有效去除噻吩类有机硫化物的同时,减少燃油中正十六烷、正辛烷、甲苯等可用非硫碳化物的损失,对吸附脱硫获取清洁油品的研究具有越来越重要的现实意义。
多孔氮化硼(porous boron nitride,p−BN)材料中的B—N 键是活性炭中C—C 键的等电子体,更重要的是,B—N键具有C—C键所不具备的局域极性,因此有望对噻吩类硫化物等极性化合物有更强的选择吸附性,从而达到优良的吸附脱硫效果。此外,p−BN 具有高的比表面积、孔隙率、耐腐蚀性和抗氧化性、高温稳定性和局部电偶极矩等优良的物理化学性能[18−20],保障了对极性分子有强吸附性能以及在恶劣环境中的应用和吸附后高温再生的可行性。
基于p−BN 优良的吸附性能以及B—N 极性成键特征,p−BN 有望对噻吩类硫化物等极性化合物有更强的选择吸附性。本文采用基于密度泛函的第一性原理计算,研究了p−BN 和含本征空位缺陷的p−BN 对燃油中噻吩类硫化物(DBT、BT、T)和主要高热能成分正十六烷(n−hexadecane)、正辛烷(n−octance)吸附和吸附选择性。通过系统的电子结构分析,揭示了p−BN 吸附脱硫机理,为设计更加高效的脱硫吸附材料及后续相关实验研究提供了理论依据。
1 计算方法与模型
1.1 计算方法
本文采用基于密度泛函理论的第一性原理计算,全部工作在Materials Studio 中DMol3模块进行[21−22]。在广义梯度近似(GGA)下执行自旋极化计算,采用Perdew−Burke−Ernzerhof(PBE)泛函描述电子的交换相关效应,并考虑了Grimme 开发的D3 色散校正(PBE+D)[23]。色散校正可更加合理地描述吸附现象,它能够同时将化学作用与范德华力引入,使密度泛函方法更加准确地计算体系能量。采用双数值轨道+p轨道极化函数的DNP基组,电子轨道截断半径为4.6 Å(1 Å=0.1 nm,后同)[21]。核处理采用最精确的全电子相对论方法,考虑了所有核电子并明确地引入相对论效应[24]。自洽场计算(SCF)的收敛阈值为0.001 eV[25]。使用Pulay 开发的直接反演迭代子空间技术加速所有体系上的SCF 收敛,子空间大小为6。所有几何优化的收敛标准均为:力梯度收敛精度为0.01 eV;位移收敛精度为0.005 Å;能量收敛精度为0.001 eV。对于结构优化和能量计算,采用了5×5×1 Monkhorst−Pack 的k 点,确保体系总能量收敛于0.01 eV。对于电子结构有关的计算,采 用 布 里 渊 区 取 样 为9×9×1 Monkhorst−Pack 的k点。
1.2 计算模型
采用周期平板模型模拟p−BN,在一个3×3×1的六方氮化硼(hexagonal boron nitride,h−BN)超胞中引入孔结构,经过完全的结构优化,形成了具有12元环孔结构的p−BN 原胞[26−27],真空层设置为18 Å,优化后晶格参数为6.83 Å,如图1所示。
在模拟吸附过程时,为了消除周期性结构之间的相互作用以及周期结构与其镜像结构之间的相互作用,建立了3×3×1 的p−BN 超胞模型,该模型由54 个B 原子和54 个N 原子组成,优化后晶格参数为20.48 Å。考虑了氮原子空位(nitrogen vacancy, VN)和硼原子空位(boron vacancy, VB)两种类型的空位缺陷,通过从p−BN超晶胞中去除单个N原子和B原子构建单原子空位缺陷,并对其进行完全的几何结构优化。
1.3 吸附能
吸附能(Eads)计算公式如下:
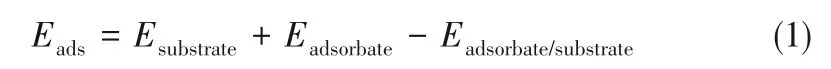
式中,Esubstrate、Eadsorbate和Eadsorbate/substrate分别是p−BN、有机分子和吸附后有机物与p−BN 超胞的总能量。根据定义,Eads>0 表示吸附是放热过程,有利于吸附反应的进行。
1.4 缺陷形成能
为探究p−BN材料中VB和VN缺陷的形成条件并分析其稳定性,使用式(2)计算不同化学气氛下VB和VN缺陷的形成能(Eform)[28−29]:

式中,Etot是具有VB或VN缺陷的p−BN 的总能量;nB和nN分别是B 和N 原子的数量;μe是电子化学势;eν 代表价带顶的能量; q 是体系的电荷。在此项研究中,只考虑电中性状态,即q = 0。因此,式(2)重写为:
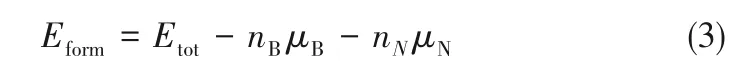
对于p−BN 的任何生长条件,μN和μB受热力学条件约束:

富硼条件(μB=μB,0;μN=μBN-μB,0)和富氮条件(μN=μN,0;μB=μBN-μN,0)代表两个极限生长条件,μB,0和μN,0分别是参考体系的化学势。由式(3)和式(4)可以计算不同化学条件下p−BN空位缺陷形成能。
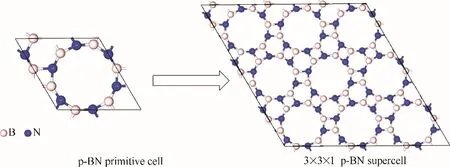
图1 p−BN超胞模型的建立Fig.1 The establishment process of the p−BN supercell
2 结果与讨论
2.1 二苯并噻吩和烷烃在p-BN上的吸附
由于DBT 分子中同时含噻吩环和苯环,本文首先重点考察了p−BN 对噻吩类含硫化合物DBT 及烷烃化合物正十六烷和正辛烷的吸附。为获取最稳定的吸附构型,分别采用平行和垂直等多种初始吸附模型将几种有机分子置于p−BN 上,优化后得到最稳定的吸附构型如图2所示。
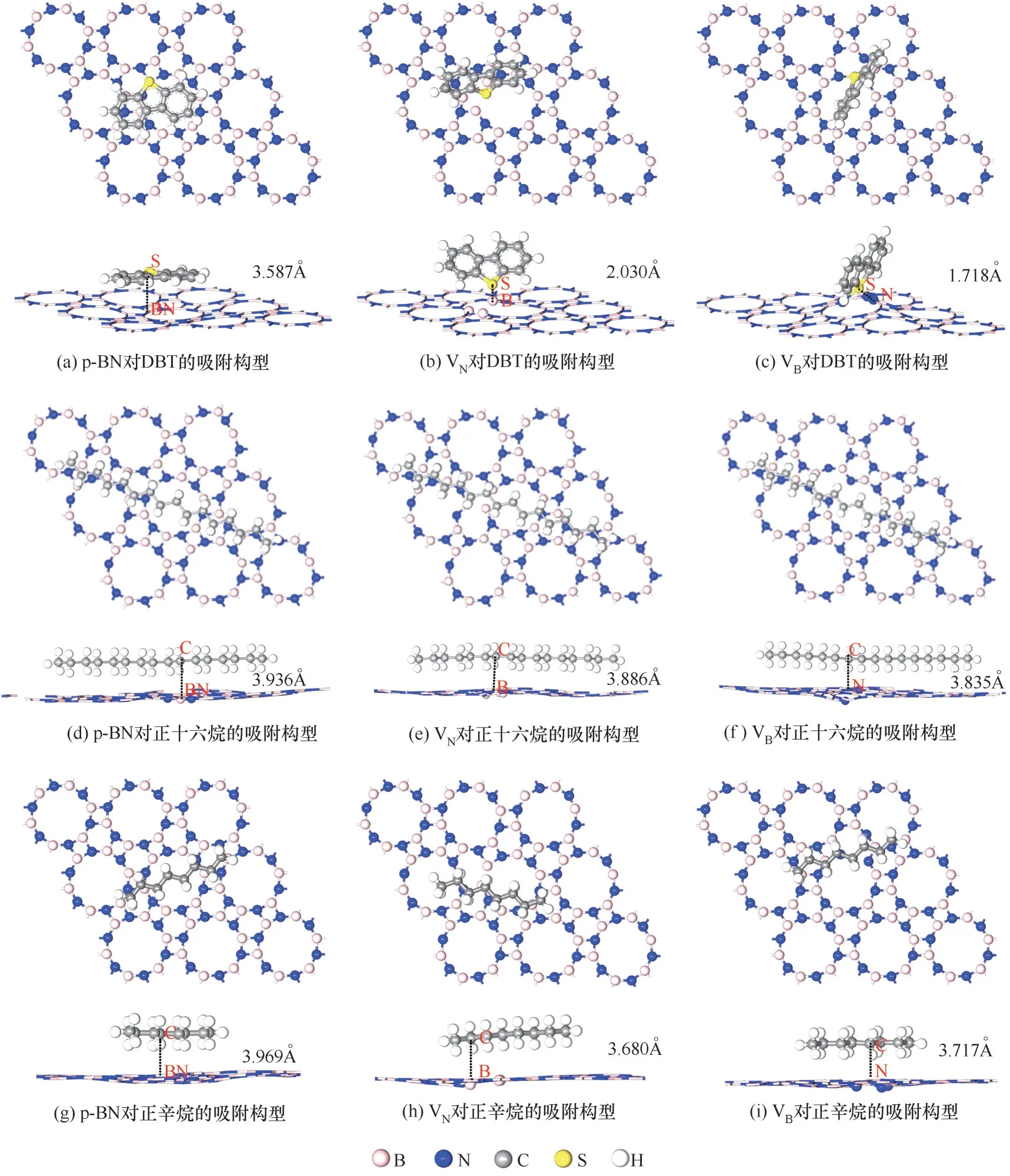
图2 p−BN、VN和VB分别对DBT、正十六烷和正辛烷最佳吸附构型的俯视及侧视图Fig.2 The geometric configurations of adsorbed DBT,n−hexadecane and n−octane on perfect p−BN,p−BN with VN defect and p−BN with VB defect,respectively
DBT 分子吸附到p−BN 后,几乎与p−BN 保持平行构型[图2(a)],与p−BN 之间的最近距离为3.587 Å。当在p−BN 上引入B、N 空位缺陷后,DBT 中的S原子一端倾斜吸附至VB和VN缺陷上[图2(b)、(c)],与缺陷周围的N 和B 原子形成N—S 和B—S 键,最短键长分别为2.030 和1.718 Å。根据吸附后N—S 和B—S 键长远小于N—S 和B—S 之间的范德华距离(3.350和3.930 Å),可以推断N—S和B—S之间形成了较强的化学相互作用。说明引入空位缺陷后增强了p−BN 与DBT之间的相互作用。正十六烷在p−BN 上的吸附如图2(d)~(f)所示,C 原子与p−BN、VN和VB之间的吸附距离分别为3.936、3.886 和3.835 Å,与范德华半径相当,呈弱相互作用。因此,与吸附正十六烷相比较,含单原子空位缺陷的p−BN 能很好地选择性吸附去除噻吩类含硫化合物DBT。此外,p−BN 对正辛烷吸附也非常弱,如图2(g)~(i)所示,三种p−BN 对正辛烷的吸附距离(3.969,3.680 和3.717 Å)也较大,呈现出一种弱吸附作用。
吸附能的结果(图3)表明,DBT、正十六烷以及正辛烷分子在p−BN 上的Eads分别为0.081,−0.130和−0.097 eV,表明完整的p−BN 对DBT 有弱相互作用,而对正十六烷和正辛烷分子的吸附热力学是不稳定的。考虑到h−BN 为层状结构,分子间力在这类材料中有重要的作用,对吸附能的计算引入了色散校正。经色散校正(PBE+D)后,p−BN 对DBT 的Eads增大到1.395 eV,与PBE 计算的结果相比增大了1.314 eV,表明p−BN 对DBT 的吸附主要是分子间力作用。对正十六烷和正辛烷的吸附,经色散校正后,Eads增加到0.600 和0.457 eV。显然,这种作用明显小于p−BN 与DBT 之间的相互作用,p−BN 对DBT与正十六烷的Eads差值依然高达0.795 eV,保障了p−BN对DBT的选择性吸附。
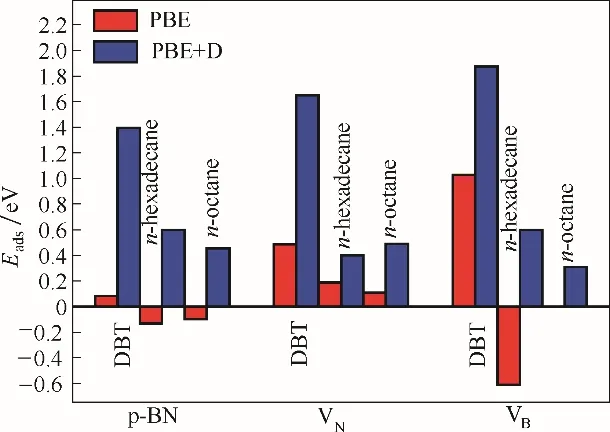
图3 完整的p−BN、VN和VB分别对DBT、正十六烷和正辛烷的吸附能(Eads)Fig.3 Calculated adsorption energies(Eads)for DBT,n−hexadecane and n−octane on the perfect p−BN,VN and VB,respectively
如图3 所示,引入VN和VB缺陷后,p−BN 对DBT的Eads显著增加,分别达到0.487 和1.028 eV。当引入色散校正后,吸附能增加到1.650 和1.875 eV,分别增加了1.163 和0.847 eV。因此,引入空位缺陷后,p−BN 与DBT 之间的分子间力和化学吸附同时增强。其中VB缺陷对DBT 的吸附以化学吸附为主,对DBT 具有更高的Eads值,大于报道[30]的DBT 在Ni−Mo−S 纳米簇上的最高吸附能值(162.14 kJ/mol,1.68 eV,GGA−RPBE,DNP)。VN和VB缺陷对正十六烷分子的吸附能分别为0.190 和−0.612 eV(加入色散矫正后的结果分别为0.400 和0.600 eV)。较小的吸附能表明正十六烷与VB和VN缺陷之间仅存在弱相互作用且主要为分子间力。更重要的是,在VN和VB缺陷上,p−BN 对DBT 和正十六烷Eads差值分别为0.297 和1.640 eV(加入色散矫正后分别为1.250 和1.275 eV),保障了材料能在去除DBT 的同时不吸附正十六烷。并且具有空位缺陷p−BN 对正辛烷的吸附也相对较弱。
2.2 p-BN吸附DBT的电子结构分析
在本文所采用的计算方法和计算水平下,计算所得p−BN 的带隙为4.121 eV[图4(a)],是一个宽带隙的半导体。这一结果与之前文献报道[26]一致,说明本文采用的计算方法和计算水平能很好地描述p−BN 的电子结构。引入VN和VB缺陷后,态密度能级出现明显的自旋分裂[图4(b)、(c)],Fermi 能级附近出现缺陷能级。在VN上[图4(b)],位于导带底的Fermi 能级附近出现一个新的半占据能级和三个未占据缺陷能级,呈n型半导体的特性;而在VB上[图4(c)],在价带顶的Fermi 附近出现一个半占据和三个未占据的缺陷能级分布,呈p 型半导体的特性。这些位于Fermi 能级附近的缺陷能级很容易获得或失去电子,因此,是影响体系化学吸附能力和催化活性的关键[27]。
为揭示缺陷p−BN 吸附脱硫的机理,系统分析了吸附后各构型的电子结构:态密度(图5)、分子轨道占据态(图6)以及电荷密度(图7)。VN吸附DBT后[图5(a)],缺陷能级发生明显的分裂,并且分态密度(partial density of states, PDOS)显示B 原子的2p轨道和S 原子的2p 轨道存在明显的共振现象[图5(c)],表明VN与DBT 之间存在的化学相互作用主要通过缺陷周围的B 与硫化物中的S 以共价键方式成键。此外,吸附后体系的HOMO 轨道[图6(a)]主要位于DBT 的S 原子与缺陷周围的B 原子上,二者发生了明显的轨道重叠现象,证实了DBT 与VN之间的化学相互作用。总电荷密度图[图7(a)]可以看出B—S键的形成;差分电荷密度图[图7(c)]中可以看出,BN和S 之间存在电荷的耗尽区域,VN缺陷周围B 原子上出现电荷富集,表明部分电荷从DBT 转移到VN上。Hirshfeld 电荷分析(表1)表明DBT 分子吸附到VN上,向VN缺 陷 位 点 周 围 的B 转 移 了0.169 e 的电荷。
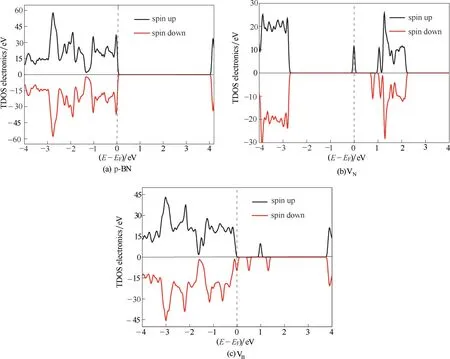
图4 p−BN、VN 和VB 的总态密度(TDOS)Fig.4 The total density of states(TDOS)for p−BN,VN and VB
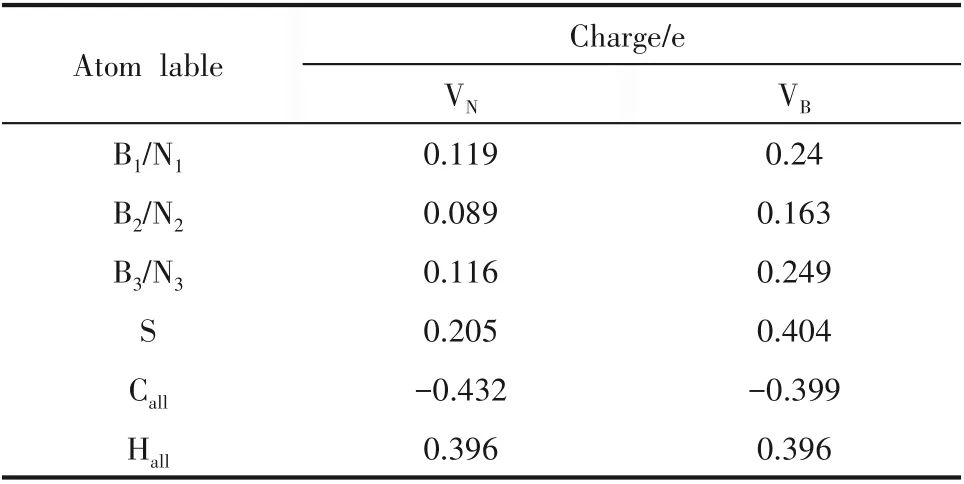
表1 DBT分别吸附在VN和VB上的Hirshfeld电荷分布Table 1 Hirshfeld atomic charge of DBT adsorption on VN and VB defect configuration
VB吸附DBT 后[图5(b)],缺陷能级远离Fermi 能级:一方面,两个缺陷能级向能量减小的方向移动,形成一个占据轨道(−0.843 eV)和一个半占据轨道,占据轨道的增加表明p−BN 获得了电子使体系能量降低;另一方面,两个缺陷能级则向高能量方向移动。这种能级的分裂使吸附体系趋向更加稳定的状态。仔细观察PDOS[图5(d)]可发现,DBT 和VB之间的成键主要是缺陷周围N原子的2p轨道和S原子的2p 轨道相互作用的结果。总电荷密度图[图7(b)]表明DBT 与VB之间有N—S 键的形成,差分电荷密度图[图7(d)]中可以看出,部分电荷从DBT转移到VB上,Hirshfeld 电荷分析进一步表明吸附的DBT 分子向VB空位附近N 原子的电子转移量为0.401 e。DBT 与VB之间更加明显的轨道成键[图5(d),图6(b)]与电子转移,证实了VB与DBT 之间存在强化学吸附作用。
2.3 燃油中其他有机物在p-BN上的吸附
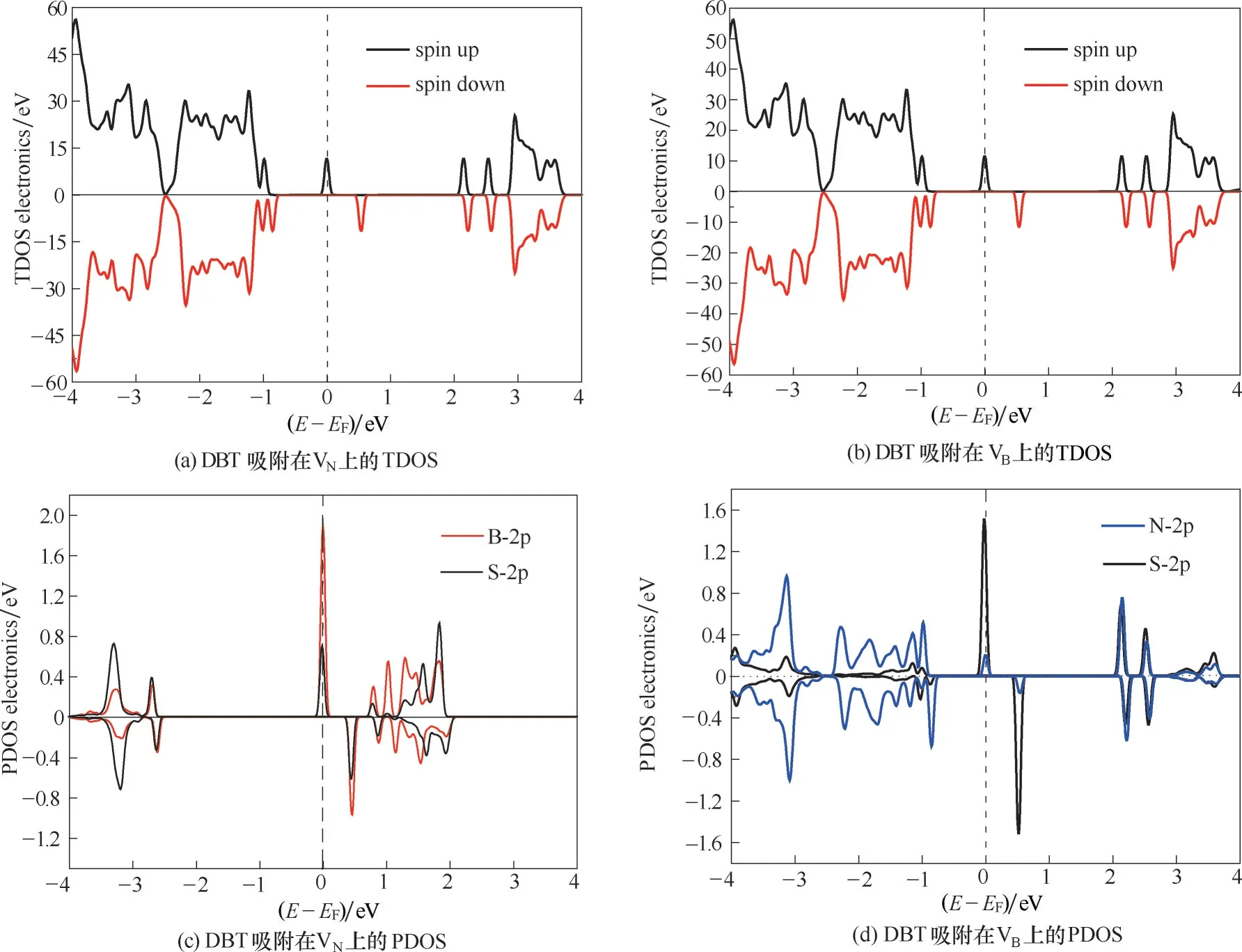
图5 DBT吸附在VN 和VB 上的总态密度(TDOS)和分态密度(PDOS)(费米能级的位置在图中用虚线标出)Fig.5 Total density of states(TDOS)and projected density of states(PDOS)for DBT adsorbed on VN and VB(the position of the Fermi level was marked in dashed line)

图6 DBT分别吸附在VN 和VB缺陷上的最高已占据分子轨道Fig.6 The highest occupied molecular orbital for DBT adsorbed on VN and VB defects
由于含VB缺陷的p−BN 能更有效地选择性吸附燃油中的DBT,接下来的计算中以含VB缺陷为例研究了p−BN与燃油中其他硫化物[9,31]如噻吩(T)、苯并噻吩(BT)、4,6−二甲基二苯并噻吩(4,6−DMDBT)和非硫化物甲苯(toluene)的相互作用。图8 为完全优化后的几何构型,其中噻吩,苯并噻吩[图8(c)、(d)]中的S 与VB空位处的三个N 原子发生强的相互作用,最短的S−N 距离分别为1.675、1.712 Å;4,6−二甲基二苯并噻吩[图8(b)]分子较大,由于S 原子周围H 原子形成的局部位阻效应使得H 原子与空位缺陷处的作用较为明显,最短的H—N 距离为2.752 Å,而S—N 距离为3.566 Å;甲苯[图8(a)]分子中的C、H 原子与VB缺陷周围的N 原子的吸附距离分别为3.164和3.382 Å。由此可见,VB对含硫有机物的吸附强于对甲苯的吸附。
如图9 所示,从吸附能的计算可以看出具有VB缺陷的p−BN 对这几种有机分子的吸附存在明显差异:VB对含硫有机分子苯并噻吩、噻吩和4,6−二甲基苯并噻吩的吸附能分别达1.791、1.374 和1.217 eV,在考虑了色散作用下吸附能分别增加到2.326、1.830 和1.615 eV;而对非硫有机物甲苯的吸附能为0.173 eV,仅为其他三种含硫化物Eads的1/7~1/10。综合前面p−BN 对DBT 和正十六烷、正辛烷的吸附研究,p−BN 特别是含VB缺陷的p−BN 对燃油中的含硫有机物有强吸附作用,而对正十六烷、正辛烷和甲苯等碳化物的吸附很弱甚至不吸附,因而能选择性去除燃油中的含硫有机物,可作为一种理想的吸附脱硫材料。
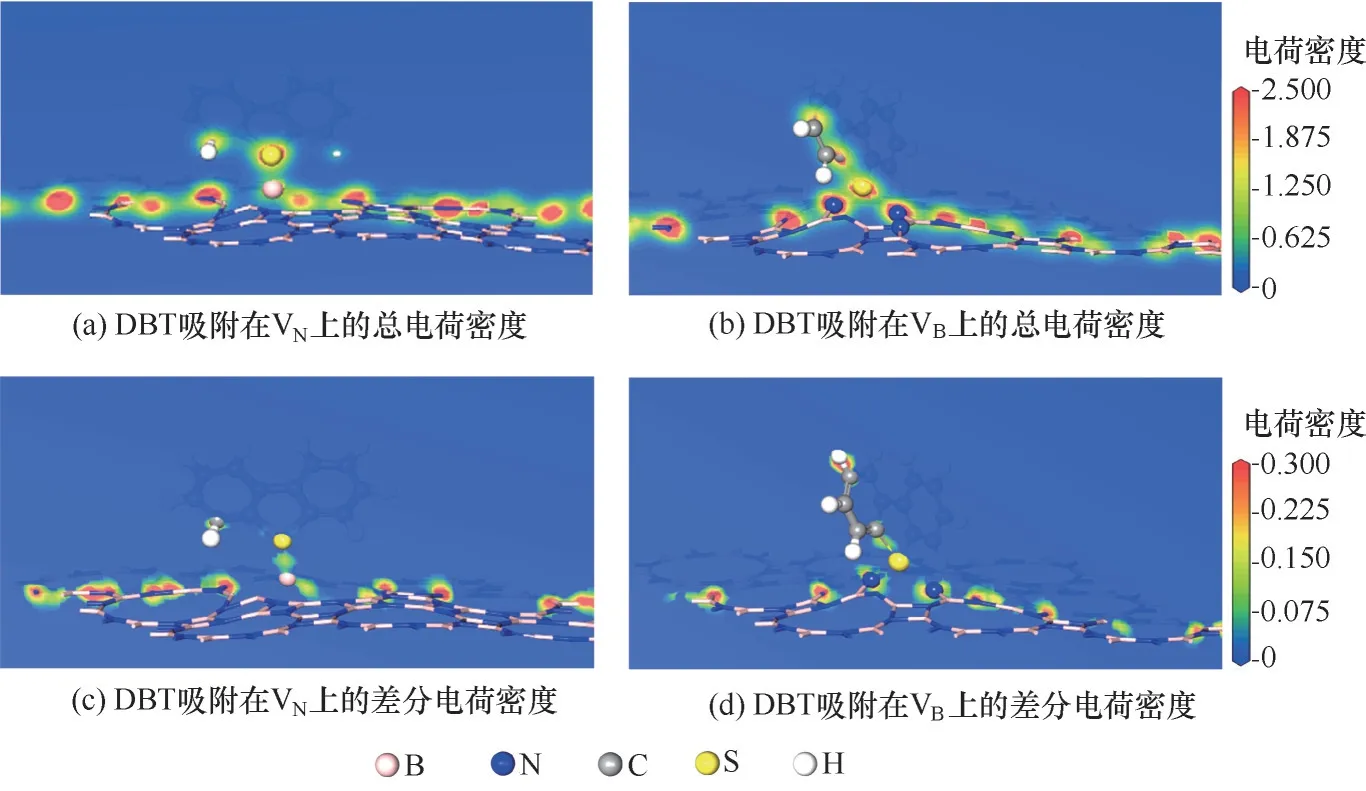
图7 DBT分别吸附在VN和VB缺陷上的总电荷密度和差分电荷密度图Fig.7 Total charge density and charge−density difference plots of DBT adsorbed on VN and VB defects,respectively
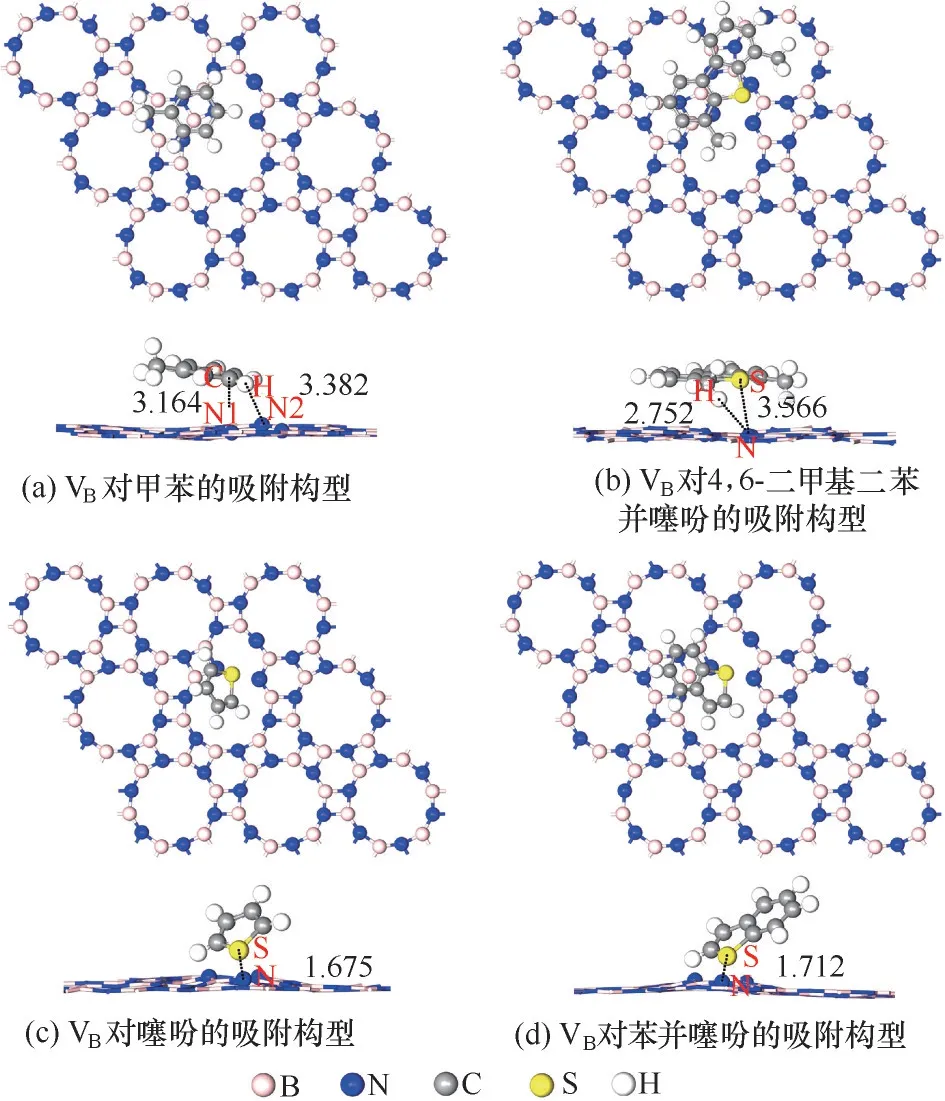
图8 甲苯(toluene)、4,6−二甲基二苯并噻吩(4,6−DMDBT)、噻吩(T)和苯并噻吩(BT)在VB上最优吸附构型Fig.8 Optimized geometric configurations of adsorbed toluene,4,6−DMDBT,T and BT on VB
2.4 缺陷p-BN的稳定性
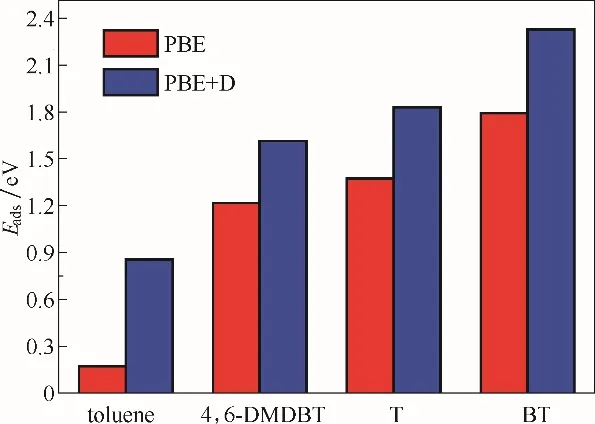
图9 甲苯、4,6−二甲基二苯并噻吩、噻吩和苯并噻吩在VB上的吸附能(Eads)Fig.9 Calculated adsorption energies(Eads)for toluene,4,6−DMDBT,T and BT on the VB,respectively
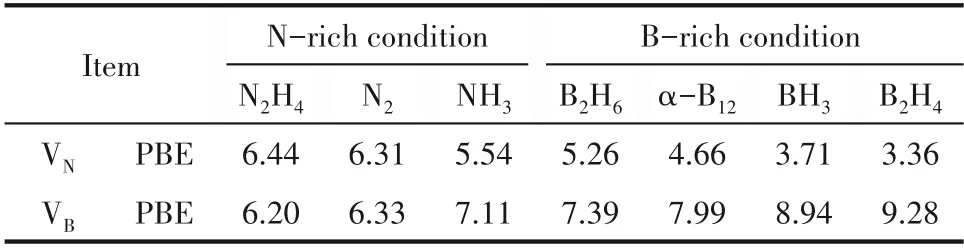
表2 在各种化学条件下VB和VN的缺陷形成能(Eform/eV)Table 2 The defect formation energies(Eform/eV)of the VB and the VN under various chemical conditions
空位缺陷的形成能依赖于p−BN 合成的化学环境,不同反应物或化学氛围(B 源和N 源)的选择将达到降低空位缺陷形成能的目的。缺陷形成能的计算结果如表2所示,在富硼环境下,VN缺陷形成能小于其在富氮环境下的形成能。例如N2H4、N2和NH3化学氛围下,VN的缺陷形成能分别为6.44、6.31和5.54 eV;而在富硼环境下(B2H4、α−B12和BH3)形成能降低至3.36、4.66 和3.71 eV。因此,在富硼尤其是B2H4环境下更有利于VN形成。而在富氮条件下VB具有较小的形成能,例如,在N2H4气氛下VB具有最低形成能为6.20 eV,表明在富氮环境下更有利于VB形成。因此,为获取具有较强吸附脱硫效果的p−BN,采用N2H4为氮源比传统的N2和NH3氛围[32]下制备的材料更加有利于VB缺陷的形成。
3 结 论
用基于DFT 的第一性原理计算系统地研究了p−BN 对DBT 等噻吩类硫化物和非硫化物正十六烷、正辛烷和甲苯的吸附行为。计算结果表明,p−BN 特别是当引入空位缺陷时可选择性去除燃油中DBT等含硫有机物;p−BN对含硫有机物的强吸附源于B—N 极性键与极性分子之间的分子间力;当引入空位缺陷后,缺陷能级与S 原子形成化学相互作用,进一步增强了对含硫有机物的吸附。基于这些研究结果,具有B、N 空位缺陷的p−BN 可以选择性除去燃油中含硫有机物。最后,计算了不同化学条件下VN、VB的缺陷形成能,揭示了空位缺陷的形成依赖于其合成氛围。该工作对于设计和合成新型吸附脱硫材料具有重要的指导意义。
