基因组测序技术解析耐除草剂转基因水稻G2-7的分子特征
2020-09-25杨江涛王旭静王志兴
马 硕 焦 悦 杨江涛 王旭静,* 王志兴,*
基因组测序技术解析耐除草剂转基因水稻G2-7的分子特征
马 硕1,**焦 悦2,**杨江涛1王旭静1,*王志兴1,*
1中国农业科学院生物技术研究所 / 农业农村部农业转基因生物安全评价(分子)重点实验室, 北京 100081;2农业农村部科技发展中心, 北京 100122
外源DNA片段的拷贝数及插入位点的侧翼序列等分子特征信息是转基因植物安全评价过程中必需要提供的信息。本研究利用基因组测序结合生物信息学对耐除草剂转基因水稻G2-7的T-DNA插入位点、拷贝数和侧翼序列进行鉴定。利用Illumina NovaSeq 6000平台对G2-7进行全基因组测序, 共获得47.13 Gb的测序数据, 通过与转基因载体和参考基因组序列的比较, 确定了G2-7中T-DNA在受体基因组中的插入位点。结果显示, 外源DNA片段以单位点单拷贝形式插入到水稻1号染色体的36,189,491~36,189,507位置, 造成水稻基因组16 bp DNA缺失, 无载体骨架的插入。同时我们获得外源基因插入位点5′侧翼序列375 bp和3′端侧翼序列353 bp, 并通过PCR扩增和Sanger测序进一步证明获得的侧翼序列是正确的。研究结果为转基因水稻G2-7的安全评价及转化体特异性检测提供了有效的数据支撑, 同时也证明全基因组测序(WGS)是解析转基因植物分子特征的有效方法。
基因组测序; 转基因水稻; 分子特征; 拷贝数; 侧翼序列
分子特征是转基因植物的身份象征, 是转基因植物安全评价的基础, 也是转基因植物检测监测的先决条件和主要依据。在我国转基因植物安全评价中, 自环境释放阶段开始, 研发者需要提供外源DNA片段在受体基因组中的拷贝数、整合位点、插入位点处的侧翼序列等方面的分子特征信息。
分析转基因植物分子特征的传统方法有多种, 其中分析基因拷贝数的常用方法有Southern杂交、荧光定量PCR和数字PCR等[1-3], 分析外源基因的整合位点和侧翼序列的常用方法为基于PCR的染色体步移技术, 包括TAIL-PCR[4-5]、反向PCR[6-7]、RSE PCR (restriction site extension PCR)[8]和接头介导的PCR (adapter ligated PCR)[9]等。这些方法虽然已在实践中得以验证和应用[10-14], 但每种方法都有其自身的优缺点和局限性, 有时很难获得理想结果。
近年来, 随着高通量测序技术的发展, 全基因组测序已成为解析转基因植物分子特征的一种新方法, 并建立起SBS (southern by sequence)、T-DNA捕获测序和全基因组测序等不同的分析转基因植物分子特征的技术体系[15-17], 用于解析转基因拟南芥[16,18]、玉米[19]、水稻[20-22]和大豆[23]中外源DNA片段在受体基因组中的整合情况及拷贝数分析, 且用此方法发现在转基因水稻[24]和玉米[25]中存在非预期插入。
G2-7为转基因的耐草甘膦水稻, 是通过农杆菌介导法将基因导入粳稻品种中花11 (ZH11)获得的独立转化事件, 具有很好的耐受草甘膦的能力[26]。目前G2-7已申请了中间试验, 通过Southern杂交证明外源基因在受体基因组中为单拷贝插入, 但通过TAIL-PCR、反向PCR等方法未能获得外源DNA片段插入位点处的侧翼序列[27]。因此, 本研究利用全基因组测序技术结合生物信息学分析, 解析外源DNA片段在受体基因组中的插入位点及其侧翼序列, 明确转基因水稻G2-7的分子特征, 为其后期的安全评价提供数据支撑。
1 材料与方法
1.1 植物材料
所用植物材料为耐草甘膦转基因水稻G2-7及其受体中花11。G2-7是通过农杆菌介导法将耐草甘膦基因导入中花11而获得, 转化时所用植物表达载体为p13UG2[26]。p13UG2质粒DNA序列作为后期测序数据的参考序列。
1.2 试验方法
1.2.1 DNA提取及全基因组测序 利用基因组提取试剂盒(北京博迈德基因技术有限公司)提取水稻叶片基因组DNA, 琼脂糖凝聚电泳对提取的基因组DNA进行质量检测。用Covaris破碎仪将基因组DNA处理成350 bp左右的DNA片段, 利用NEB Next Ultra DNA Library Prep 试剂盒进行建库, 并用NGS3K/Caliper对文库的DNA片段大小进行检测, 用qPCR方法对文库的有效浓度(3×10–9mol L–1)进行准确定量。本研究共构建了G2-7、中花11 (ZH11)和中花11+1拷贝质粒DNA (ZH11-p) 3个基因组文库。构建好的文库采用PE150双末端测序法在Illuminia NovaSeq 6000平台进行测序, 测序深度为70×。
1.2.2 测序数据质量控制及比对 分析测序原始数据碱基错误率, 并对测序原始数据(raw reads)进行过滤(去除带接头、单端测序read中N的比例大于10%、以及单端测序read中含有的低质量碱基数超过其长度比例50%的reads)处理获得clean reads。
利用BWA-0.7.17软件, 经MEM算法将Clean reads与质粒DNA序列进行比对。比对分析后, 将clean reads分成3组, 即完全与参考基因组序列匹配的reads, 完全与质粒DNA序列匹配的reads, 结合区(部分与质粒DNA序列匹配, 部分与基因组序列匹配)序列的reads。匹配到质粒DNA和结合区的reads将用于后期外源基因拷贝数、整合位点及侧翼序列分析。
1.2.3 外源基因拷贝数、整合位点及侧翼序列分析
将匹配到质粒DNA和结合区的reads通过IGV-2.5.0 (integrative genomic viewer)进行可视化分析, 通过分析结合位点的数量来确定外源DNA插入的拷贝数, 通过与质粒DNA比对分析是否有载体骨架插入, 其中ZH11+P和ZH11测序数据分别作为阳性对照和阴性对照。
接合区reads物理位置指示了T-DNA在受体基因组上的整合位点。编写Python脚本对接合区序列进行提取, 使用SOAPdenovo进行侧翼序列拼接, 拼接完后与转化载体p13UG2序列和日本晴(L. spp.,var.) 参考基因组进行分别比对, 获得外源基因在受体基因组中整合位点及侧翼序列。
1.2.4 转基因水稻G2-7分子特征验证 根据基因组测序获得的T-DNA 5′端和3′端侧翼序列设计引物5F (5′-GGTGGCTGGGCGATGTGC-3′)和3F (5′- ACTTCAAACAAGTGTGACAA-3′), 根据T-DNA LB端和RB端序列设计引物5R (5′-GTACTCGCCG ATAGTGGAAACCG-3′)和3R (5′-CATTGTCAAATC ATAGAGCAATT-3′)。以G2-7叶片DNA为模板, 分别以5F/5R和3F/3R为引物对进行PCR扩增。PCR反应程序: 为95℃预变性8 min; 95℃ 30 s, 54℃ 30 s, 72℃ 1 min, 35个循环; 72℃延伸5 min。PCR扩增得到的DNA片段进行Sanger测序, 并与全基因组测序获得的侧翼序列进行比对分析。
2 结果与分析
2.1 全基因组测序数据分析
Illumina NovaSeq 6000高通量测序得到的原始图像数据文件经CASAVA碱基识别(base calling)分析转化为原始测序序列(raw bases)。对获得的原始测序数据进行质量控制, 过滤掉带接头(adapter)的读序、单端测序读序中N数量超过此读序长度比例10%的读序、及单端测序读序中含有的低质量(Q≤5)碱基数超过该条读序长度比例50%的等低质量读序, 获得Clean Bases和Clean Reads。本研究获得样品的原始测序量为41.16~47.13 G, 有效读序为274,371,776~314,171,200, Q30大于90%, 说明获得的测序数据丰富可靠(表1)。

表1 高通量测序数据质量控制统计
Q20、Q30分别代表Phred数值大于20、30的碱基占总体碱基的百分比, 表示的碱基正确识别率为99.0%和99.9%。
Q20 and Q30 represent the percentage of bases with Phred value more than 20 and 30 in the total base, respectively, indicating that the correct recognition rate of bases is 99.0% and 99.9%.
2.2 外源基因拷贝数分析
利用BWA-0.7.17软件将获得clean reads与质粒DNA序列进行比对, 获得了比对到载体骨架、T-DNA和结合区的DNA读序(表2)。分析发现, G2-7中匹配到结合区的读序有348条, 其中匹配到3′端结合区的读序有156条, 匹配到5′端结合区的读序有192条, 外源DNA片段与受体基因组的结合位点为2个(图1); G2-7中完全匹配到T-DNA区的读序有6130条, ZH11-p中完全匹配到T-DNA区的读序有10,487条(图2)。以上结果说明, 外源DNA在受体基因组中为单位点单拷贝插入。
为了明确是否有载体骨架插入, 本研究分析测序数据与载体骨架序列的匹配情况, 发现G2-7中匹配到载体骨架的读序有3条, 分别定位在载体参考序列的171~233、4576~4725和4586~4735位置, ZH11-p中完全匹配到载体骨架区的读序有35,163条, ZH11中匹配到载体骨架上的读序为0。对载体骨架4483~5036位置设计引物对进行PCR扩增, 在G2-7水稻中未见该段序列存在(图3)。且G2-7与单拷贝对照ZH11-p在该区域的测序深度相差非常大, 由此认为G2-7中无载体骨架的插入, G2-7中匹配到载体骨架上的读序为污染所致。
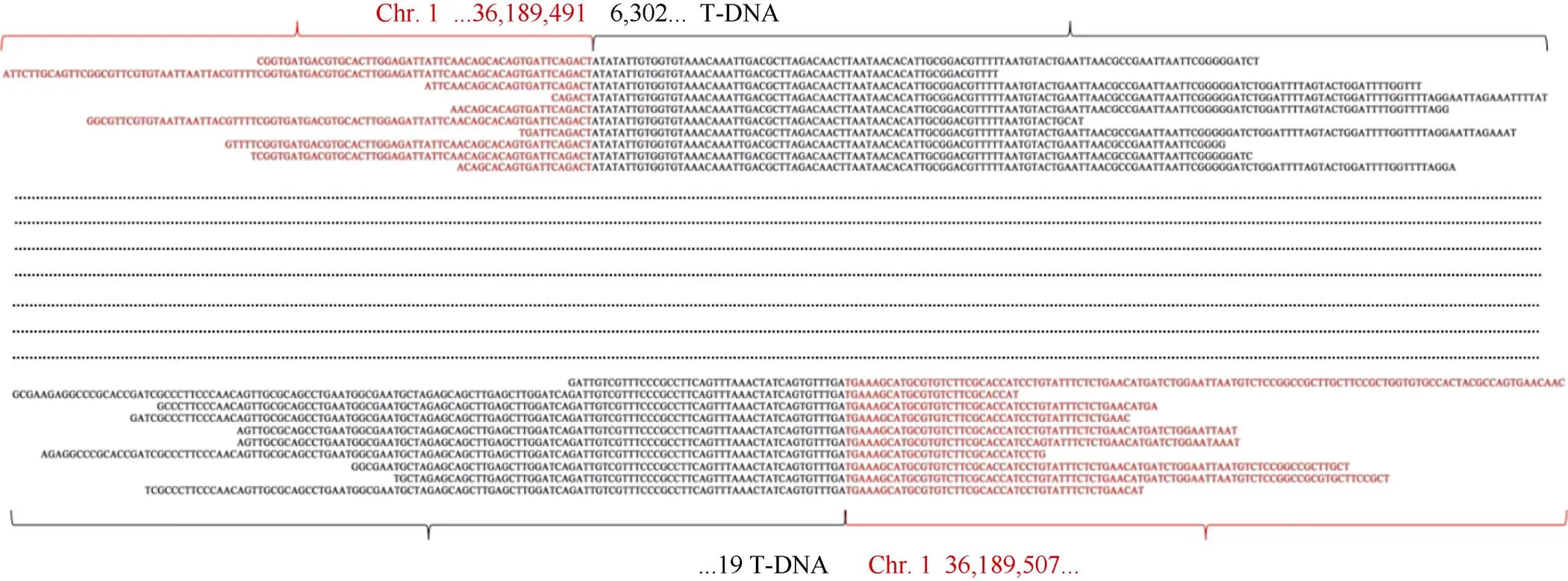
图1 G2-7中外源插入片段与受体基因组结合位点分析(部分结合区序列的比对结果)

图2 测序数据与质粒DNA比对结果的可视化
2.3 外源基因在受体基因组中的整合位点及侧翼序列分析
将G2-7中匹配到结合区序列用SOAPdenovo进行拼接, 获得了插入位点处3′端接合区序列780 bp和5′端接合区序列823 bp。其中, 3′端接合区有353 bp为水稻基因组序列, 有427 bp为T-DNA序列, T-DNA序列在3′端缺失42 bp; 5′端接合区序列有375 bp为水稻基因组, 有448 bp为T-DNA序列, T-DNA序列在5′端缺失7 bp。通过与已知的水稻基因组序列进行Blast比对分析, 确定T-DNA插入到受体基因组的1号染色体36,189,491~36,189,507 bp处, 在T-DNA与受体基因组整合过程中造成16 bp基因组DNA缺失(图4)。

图3 载体骨架匹配读序的PCR验证
1: G2-7; 2: ZH11; 3: p13UG2.

图4 外源DNA片段在受体基因组中的整合位点及侧翼序列分析
A: G2-7转化体侧翼序列和整合位点分析; B: G2-7转化体插入序列整合情况示意图。
A: analysis of flanking sequence and integration site of G2-7; B: sketch map of insert DNA integration in G2-7.
2.4 侧翼序列的Sanger测序验证
根据全基因组测序获得的5′端侧翼序列和T-DNA的LB端序列, 3′端侧翼序列和T-DNA的RB端序列设计2对引物, 以G2-7叶片基因组DNA为模板进行PCR扩增, 结果得到预期目标DNA条带, Sanger测序证明获得的3′和5′段侧翼序列与全基因组测序获得的序列一致(图5)。
3 讨论
伴随大数据时代的到来, 全基因组测序已成为解析转基因植物分子特征的一种新技术。与传统转基因植物分子特征解析方法(如Southern杂交和染色体步移技术等)相比, 全基因组测序不但具有高通量、标准化程度高、灵敏度高、重复性好和准确度高等特点, 而且能够提供插入的DNA序列信息、小片段DNA的非预期插入和DNA重排等信息, 如SBS技术能检测到50 bp小片段DNA在玉米基因组的插入[15], 全基因组测序技术能检测到100 bp 单拷贝DNA片段在玉米基因组的插入[28]。本研究在利用反向PCR、TAIL-PCR等方法无法获取到G2-7转基因水稻中外源插入序列两端的侧翼序列的情况下, 通过高通量全基因组测序技术结合生物信息学分析, 发现G2-7中外源DNA片段以单拷贝形式整合到水稻1号染色体上, 无载体骨架插入、无DNA重排, 并获得了外源DNA片段在受体基因组中插入位置的侧翼序列, 从而成功解析G2-7的分子特征。

图5 PCR电泳图及序列比对
A: 水稻G2-7转化体侧翼序列扩增; 1: G2-7-5F/5R; 2: ZH11-5F/5R; 3: G2-7-3F/3R; 4: ZH11-3F/3R。B: G2-7 5′端序列比对验证。C: G2-7 3′端序列比对验证。
A: amplification of rice transformant G2-7 flanking sequence; 1: G2-7-5F/5R; 2: ZH11-5F/5R; 3: G2-7-3F/3R; 4: ZH11-3F/3R. B: G2-7 5′ end sequence comparison and verification. C: G2-7 5′ end sequence comparison and verification.
数据分析过程中, 比对到载体骨架序列的读序可能来自于遗传转化过程中造成的载体骨架插入, 或者是受体基因组中同源序列和建库过程中细菌质粒及其他污染所造成的假阳性[15,29]。建库过程中细菌质粒或其他因素污染所造成的假阳性现象非常普遍, 而且难以完全避免[17]。判断比对到载体骨架的序列是否为假阳性, 可以通过分析这些读序的测序深度和覆盖度, 及将这些读序与受体基因组序列和已知的污染源进行比对来确定[2]。在本研究中, 将G2-7测序数据与载体序列进行比对, 发现有少量读序比对到载体骨架上, 这些读序的测序深度很低, 将非转基因水稻中花11的基因组序列与载体序列进行比对时未发现有读序比对到载体骨架上(图4), 结合PCR验证, 认为比对到载体骨架上的少量读序不是来自于载体骨架真正插入, 而是来源于建库过程中细菌质粒或其他因素污染所造成的假阳性。
在将G2-7测序数据与载体序列进行比对过程中, 发现比对到T-DNA 7800~8400位置的读序很少, 测序深度比较低, 出现gap现象, 推测可能是由于建库过程中此段DNA片段缺失或测序因素造成。为了对此进行验证, 本研究根据T-DNA的相关序列设计引物, 以G2-7叶片DNA为模板进行PCR, 经sanger测序后证明实际插入到受体基因组中的片段此区段并未缺失, 因此我们认为利用基因组测序分析外源插入片段的完整性时, 建议高通量测序与Sanger测序结合使用, 以保证结果的可靠性。
4 结论
利用高通量基因组测序技术结合生物信息学分析, 明确耐除草剂转基因水稻G2-7中外源DNA片段以单位点单拷贝的形式插入到水稻基因组1号染色体36,189,491~36,189,507 bp处, 造成16 bp水稻基因组DNA缺失, 获得外源插入片段3′端侧翼序列353 bp, 5′端侧翼序列375 bp。本研究结果为G2-7商业化过程中的安全评价和转化事件特异性PCR检测方法的建立提供了数据支撑。
[1] Southern E M. Detection of specific sequences among DNA fragments separated by gel electrophoresis., 1975, 98: 503–517.
[2] Yang L T, Ding J Y, Zhang C M, Jia J W, Weng H B, Liu W X, Zhang D B. Estimating the copy number of transgenes in transformed rice by real-time quantitative PCR., 2005, 23: 759–763.
[3] 姜羽, 胡佳莹, 杨立桃. 利用微滴数字PCR分析转基因生物外源基因拷贝数. 农业生物技术学报, 2014, 22: 1298–1305. Jiang Y, Hu J Y, Yang L T. Estimating the exogenous genes copy number of genetically modified organisms by droplet digital PCR., 2014, 22: 1298–1305 (in Chinese with English abstract)
[4] Liu Y G, Whittier R F. Thermal asymmetric interlaced PCR: automatable amplification and sequencing of insert end fragments from P1 and YAC clones for chromosome walking., 1995, 25: 674–681.
[5] Singer T, Burke E. High-throughput TAIL-PCR as a tool to identify DNA flanking insertions., 2003, 236: 241–272.
[6] Yan Y X, An C C, Li L, Gu J Y, Tan G H, Chen Z L. T-linker-specific ligation PCR (T-linker PCR): an advanced PCR technique for chromosome walking or for isolation of tagged DNA ends., 2003, 31: 1–7.
[7] Rosenthal A, Jones D S. Genomic walking and sequencing by oligo cassette mediated polymerase chain reaction., 1990, 18: 3095–3096.
[8] Ji J B, Braam J. Restriction site extension PCR: a novel method for high-throughput characterization of tagged DNA fragments and genome walking., 2010, 5: e10577.
[9] O’Malley R C, Ecker J R. Linking genotype to phenotype using theunimutant collection., 2010, 61: 928–940.
[10] Yang L, Xu S, Pan A, Yin C, Zhang K, Wang Z. Event specific qualitative and quantitative polymerase chain reaction detection of genetically modified MON863 maize based on the 50-transgene integration sequence., 2005, 53, 9312–9318.
[11] Windels P, Taverniers I, Depicker A, Van Bockstaele E, De Loose M. Characterization of the roundup ready soybean insert., 2011, 213: 107–112.
[12] Akritidis P, Pasentsis K, Tsaftaris A S,Mylona P V, Polidoros A N. Identification of unknown genetically modified material admixed in conventional cotton seed and development of an event-specific detection method., 2008, 11: 76–83.
[13] Wang X B, Jiang L X, Wei L, Liu L, Lu W, Li W X. Integration and insertion site of EPSPs gene on the soybean genome in genetically modified glyphosate-resistant soybean., 2010, 36: 365–375.
[14] Marie-Alice F, Philippe H, Isabel T, Marc D L, Dieter D, Roosens N H. Current and new approaches in GMO detection: challenges and solutions., 2015, 392872.
[15] Zastrow-Hayes G M, Lin H N, Sigmund A L, Hoffman J L, Alarcon C M, Hayes K R. Southern-by-sequencing: a robust screening approach for molecular characterization of genetically modified crops., 2015, 8: 1–15.
[16] Inagaki S, Henry I M, Lieberman M C, Comai L. High-through put analysis of T-DNA location and structure using sequence capture., 2015, 10:e0139672.
[17] Kovalic D, Garnaat C, Guo L, Yan Y P, Groat J, Silvanovich A. The use of next generation sequencing and junction sequence analysis bioinformatics to achieve molecular characterization of crops improved through modern biotechnology., 2012, 5: 3.
[18] Lepage E, Zampini E, Boyle B, Brisson N. Time and cost- efficient identification of T-DNA insertion sites through targeted genomic sequencing., 2013, 8: e70912.
[19] Rosalind W C, Nicholas S, Susan B, Tiffany K, David B S, Rita A M. Use of Illumina sequencing to identify transposon insertion underlying mutant phenotypes in high-copy Mutator lines of maize., 2010, 63: 167–177.
[20] Wahler D, Schauser L, Bendiek J, Grohmann L. Next-generation sequencing as a tool for detailed molecular characterization of genomic insertions and flanking regions in genetically modified plants: a pilot study using a rice event unauthorized in the EU., 2013, 6: 1718–1727.
[21] Park D, Kim D G, Jang G, Lim J S, Shin Y J, Kin J. Efficiency to discovery transgenic loci in GM rice using next generation sequencing whole genome-sequencing., 2015, 13: 81–85.
[22] Park D, Park S H, Ban Y W, Kim Y S, Park K C, Kim N S. A bioinformatics approach for identifying transgene insertion sites using whole genome sequencing data., 2017, 17: 67.
[23] Guo B F, Guo Y, Hong H L, Qiu L J. Identification of genomic insertion and flanking sequence of G2-EPSPS and GAT transgenes in soybean using whole genome sequencing method., 2016, 7: 1009.
[24] Yang L, Wang C, Holst-Jensen A, Morisset D, Lin Y, Zhang D. Characterization of GM events by insert knowledge adapted re-sequencing approaches., 2013, 3: 1–9.
[25] Siddique K, Wei J, Li R, Zhang D, Shi J. Identification of T-DNA insertion site and flanking sequence of a genetically modified maize event IE09S034 using next-generation sequencing technology., 2019, 61: 694–702.
[26] Dong Y, Jin X, Tang Q, Zhang X, Yang J, Liu X, Cai J, Zhang X, Wang X, Wang Z. Development and event-specific detection of transgenic glyphosate-resistant rice expressing thegene., 2017, 8: 885.
[27] 董玉凤. 转基因抗草甘膦水稻的获得及G2-EPSPS蛋白拆分重组后的草甘膦抗性分析. 中国农业科学院博士学位论文, 北京, 2016. Dong Y F. Development of Glyphosate-resistance Rice withand the Assessment of Reassembled G2-EPSPS after Splitted. PhD Dissertation of Chinese Academy of Agricultural Sciences, Beijing, China, 2016.
[28] Cade R, Burgin K, Schilling K, Lee T J, Ngam P, Devitt N, Fajardo D. Evaluation of whole genome sequencing and an insertion site characterization method for molecular characterization of GM maize., 2018, 6: 1–14.
[29] Lusk R W. Diverse and widespread contamination evident in the unmapped depths of high throughput sequencing data., 2014, 9: e110808.
Molecular characterization identification by genome sequencing of transgenic glyphosate-tolerant rice G2-7
MA Shuo1,**, JIAO Yue2,**, YANG Jiang-Tao1, WANG Xu-Jing1,*, and WANG Zhi-Xing1,*
1Biotechnology Research Institute, Chinese Academy of Agricultural Sciences / MARA Key Laboratory on Safety Assessment Molecular of Agri-GMO, Beijing 100081, China;2Development Center for Science and Technology / MARA, Beijing 100122, China
Molecular characterization, such as copy number and flanking sequence of foreign DNA fragment insertion site, is the important identity information, provided during safety assessment of genetic modified crop. In this study, the T-DNA insertion site, copy number and flanking sequences were identified in transgenic glyphosate-tolerant rice G2-7 based on whole genome sequencing in combination bioinformatics analysis method. 47.13 Gb clean sequence data for G2-7 was generated on Illumina NovaSeq 6000 platform. The junction reads mapped to boundaries of T-DNA and flanking sequences in G2-7 were identified by comparing with sequence of transformation vector and rice reference genome. The results showed that exogenous T-DNA fragments was integrated in the position of Chr. 1 36,189,491–36,189,507 with a single copy, 16 bp rice genome sequence was deleted at the insertion site and no insertion of vector backbone. 375 bp and 353 bp flanking host DNA sequence of 5′-end and 3′-end of the insertion DNA fragment were also obtained, respectively. The putative insertion location and flanking sequences were further confirmed by PCR amplification and Sanger sequencing. The results not only provided data support for safety assessment and event specific detection, but also demonstrated that WGS was an effective technique for identifying molecular characterization in rice.
genome sequencing; transgenic rice; molecular characterization; copy number; flanking sequence
10.3724/SP.J.1006.2020.02002
本研究由国家转基因新品种培育重大专项(2016ZX08010-003)资助。
This study was supported by the National Major Project for Developing New GM Crops (2016ZX08010-003).
王志兴, E-mail: wangcotton@126.com; 王旭静, E-mail: wangxujing@caas.cn
**同等贡献(Contributed equally to this work)
马硕, E-mail: mashuo0801@163.com; 焦悦, E-mail: jiaoyue@agri.gov.cn
2020-01-14;
2020-06-22.
URL: https://kns.cnki.net/kcms/detail/11.1809.S.20200622.1349.012.html
