葡萄糖结合蛋白(GBP)构象变化的研究及讨论
2020-08-23崔兆宁李义武丽达刘雄飞陶进崔兆宁佟毅
崔兆宁 李义 武丽达 刘雄飞 陶进 崔兆宁 佟毅


摘 要:葡萄糖结合蛋白(D-glucose Binding Protein,简称GBP)属于一类在细胞膜上起化学活性物质运载相关作用的周质结合蛋白(PBPs)。未结合葡萄糖分子的GBP (空配体GBP或“apo GBP”)的构象动力学在配体结合过程中起着至关重要的作用,了解这一过程对于设计该蛋白的抑制剂或突变具有重要意义。利用空配体GBP的全原子分子动力学模拟,确定了空配体GBP的多种亚稳态构象,模拟结果揭示了场域静电斥力或许是空配体GBP构象动力学的关键决定因素。GBP上下两块区域的蛋白结构域相互作用,与铰链区域施加的几何约束作用相平衡。在溶液中,区域相互作用导致了空配体GBP开口构象和闭口构象之间可以快速转化。此外,分子动力学模拟所绘制的空配体GBP的亚稳态与之前的荧光研究一致,在核磁共振(NMR)实验中也观察到了不同构象之间的快速平衡。结果表明,现有的晶体结构可能不代表溶液中的主要构象。
关 键 词:葡萄糖结合蛋白;分子动力学模拟;亚稳态;构象变化
中图分类号:Q503 文献标识码: A 文章编号: 1671-0460(2020)07-1409-05
Research and Discussion on Conformational Dynamics
of D-Glucose Binding Protein (GBP)
CUI Zhao-ning1, LI Yi2, WU Li-da2, LIU Xiong-fei3, TAO Jin2,[崔兆宁3] TONG Yi1*
- COFCO Biotechnology Co., Ltd., Beijing 100005, China;
2. Jilin COFCO Biochemical Co., Ltd., Jilin Changchun 130033, China;
3. Mengniu Dairy Technology (Beijing) Co., Ltd., Beijing 101107, China[崔兆宁4] )
Abstract: The D-glucose binding protein (GBP) belongs to a subclass of periplasmic binding proteins (PBPs) associated with membrane complexes for transport and chemotaxis. The conformational dynamics of glucose-free GBP (apo GBP) plays a vital role in the ligand binding process, understanding of which is important for the design of inhibitors or mutations of the protein. We have constructed molecular dynamics (MD) simulations from all-atom molecular dynamics simulations of apo GBP and have identified multiple meta-stable conformational states of the apo GBP. Our results suggest that domain-domain electrostatic repulsion is a key determinant to the conformational dynamics of apo GBP. In balance with the geometric restrains imposed by the hinge loops, domain-domain interactions result in a rapid equilibrium between the open and closed conformations in solution. Moreover, the metastable states mapped out by MD simulation for apo GBP agrees with previous fluorescence study and the rapid equilibrium between different metastable conformations has also been observed in NMR experiment. Our results suggest that the existing crystal structures may not represent the dominant conformations in solution.
Key words: Glucose binding protein; Molecular dynamics simulation; Metastable state; Conformational change
周质结合蛋白(PBPs)是一个大的受体家族。它们的关键功能包括识别配体,如小分子和离子,并将它们穿过细胞膜进行后续的生物過程。大肠杆菌葡萄糖结合蛋白(GBP)是PBPs的一个亚类[1]。基于葡萄糖浓度对分子构象动态的影响,GBP有可能成为糖尿病患者的生物传感器[2-4]。
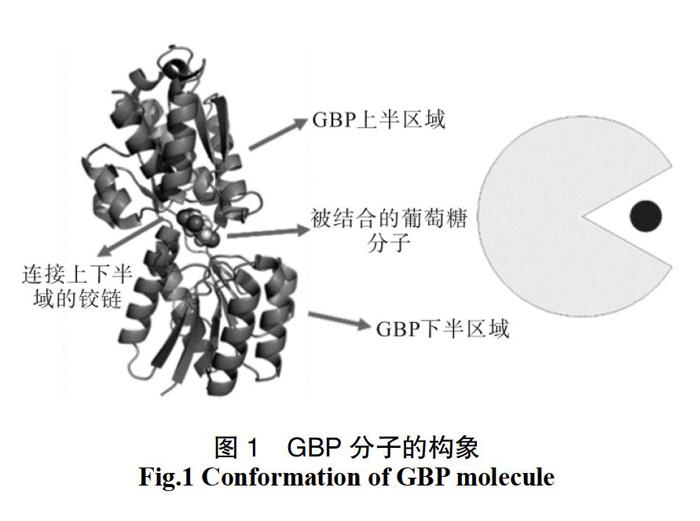
GBP由两个蛋白结构域通过铰链区域连接而成,在这两个域之间的界面上有糖结合位点[5-6]。没有葡萄糖结合的GBP构象统称为空配体构象或apo构象,而结合了葡萄糖的GBP则被称为配体构象或holo构象。如图1所示,GBP分子由上下两片区域构成,中间以铰链连接,整个分子可以是开口构象,也可以是闭口构象,与“吃豆人(Pacman)”的行为方式比较类似。不同的是,GBP上下两片区域还可以相对扭动,故GBP的不同构象可以用不同的开口角度和扭转角度2个参数加以区分(如图3)。
在GBP与葡萄糖分子的结合过程中,有两种可能的结合路径,如图2所示:(1)空配体GBP分子自身运动到合适的构象,而后葡萄糖与此构象直接结合,形成配体构象;(2)空配体GBP分子在某一个构象与葡萄糖分子结合,而后葡萄糖分子诱导GBP分子继续进行构象变化,最终抵达稳定的配体构象状态。而GBP与配体结合的过程究竟属于哪种,或是两者皆有,目前尚不完全清楚。
1 研究背景
有研究表明,GBP与葡萄糖的结合是上下两片蛋白区域逐渐闭口、葡萄糖分子逐渐嵌入的过程。此结合过程的构象变化被认为是受构象选择机制控制的,其中一个预先存在的构象被“选择”,并在配体(葡萄糖)结合后稳定下来。通过实验观察,得到了配体闭口(holo-closed)构象和空配体闭口构象(apo-closed)的X射线晶体结构。然而,最近的核磁共振(NMR)波谱研究获得了有争议的结果,表明没有单一的X射线晶体结构与核磁共振(NMR)残基偶极耦合(RDC)数据一致[7-8]。重要的是,核磁共振(NMR)实验表明,作为支撑构象选择机制的关键构象,闭口构象在溶液中可能不稳定[9]。因此,根据核磁共振(NMR)所示,溶液中可能存在多个亚稳态构象,然而,对于配体后续构象选择,在溶液的结构系综中是否存在足够百分比的空配体闭口构象尚不清楚。
在决定GBP和其他PBPs构象动态的各种因素中,铰链区域被认为是最重要的因素之一[10]。特别是Borrok M.J.等在大肠杆菌GBP的开口与闭口晶体结构之间的三段铰链区观察到显著的扭转角变化。此外,Ortega G.等研究发现,在第一个铰链节上出现了G109A/T110S双突变体,该突变体可以极大地改变蛋白质的构象动力学,使开口构象更为理想[9]。
然而,仅靠铰链区不足以解释一些诱变实验结果。近年来,一些GBP突变体,如E149C[11]、 A213R[4]、E149C/A213S/L238S[11],被用来削弱GBP的结合亲和力,以接近生理相关的葡萄糖浓度。许多突变残基既不影响铰链区,也不显著改变蛋白质-配体相互作用。为什么这些突变体能敏感地影响葡萄糖结合亲和力还没有完全弄清楚[4, 11-12]。
除了铰链区域外,蛋白结构域相互作用也可能在双域PBPs的构象动力学中发挥重要作用。在1999年,Hasemann C.A.等研究了TroA(也是PBPs中的一种)中显著的亲水蛋白结构域相互作用[13]。这种沿着接触面的相互作用使蛋白质打开结合位点进行配体交换。Nussinov R.等进一步对PBPs铰链弯曲转变过程中的蛋白结构域相互作用进行了广泛的研究,结果表明,PBPs的铰链弯曲运动可以通过改变域间的非极性埋藏表面积或引入域间静电相互作用来调节[14]。此外,在特殊情況下,域间疏水相互作用甚至可能导致多域蛋白迅速折叠。然而,很少有人研究蛋白结构域相互作用在GBP构象动力学中的作用[15]。
在本研究中,利用溶剂中未结合葡萄糖的GBP全原子分子动力学(MD)模拟,并建立了马尔可夫状态模型(MSMs),以确定亚稳态构象和它们之间的构象变化动力学。结果表明,在亚微秒的时间内,场域排斥相互作用对这两个亚稳态构象在不同开口角度和不同扭转角度下的快速平衡起着重要的调节作用。此外,铰链区域也是必不可少的,它们之间提供了必要的几何约束,以平衡各种亚稳态构象的蛋白结构域间排斥作用。研究表明,GBP的晶体构象可能在溶液中不稳定,只是因为晶体堆积效应才表现出在晶体中稳定的性质。
2 静电束缚自由能的计算
2.1 热力学循环
为了研究GBP上下蛋白结构域之间的场域静电相互作用,使用了T. Wang等报道的两个热力学循环的方案[16]。首先,敲除3个铰链环,将整个蛋白质分成上下两部分。我们删除的铰链节段为残留物110~112(节段I)、255~257(节段II)、293~295(节段III),下半部分与上半部分之间的静电结合自由能可表示为:
其中E为两片区域在溶液中结合并完成时的静电相互作用势能, 。 为上半部分在下半部分各原子位置所产生的静电势, 为下半部分的原子电荷。 和 分别为下半部分和上半部分的静电脱溶自由能,定义为溶剂与蛋白质结合时静电相互作用能的损失。
2.2 自适应玻尔兹曼解算程序(APBS)
本研究采用自适应玻尔兹曼解算程序(APBS)计算了脱溶剂化自由能和静电相互作用能[17]。APBS是一个通过数值求解泊松-玻尔兹曼方程(PBE)来模拟生物分子解的软件包:
PBE是一个二阶非线性椭圆形偏微分方程,包括静电势 、蛋白质和溶剂的介电特性 、溶液的离子强度和离子进入蛋白质内部的可达性 、蛋白质原子部分电荷的分布 。PBE常被线性化的近似为LPBE,如果假设 。
在这里,在LPBE上运算,并实现了手动配置的多网格泊松-玻尔兹曼计算,这是一个标准的单点多网格PBE计算,没有聚焦细化。单位网格大小为0.3×0.3×0.3 ?,立方体粗离子化个数为289×257× 257,其中心在Asn256 氨基酸Cα原子的位置。采用“单Debye-Huckel”边界条件求解LPBE。GBP和溶剂的介电常数分别设置为2.00和78.54。介电常数和离子可及性系数的定义采用“分子”表面定义和谐波均相平滑处理[18]。采用晶格离散化方法,将生物分子点电荷映射到最近和相邻的网格点上。溶剂分子和离子的半径分别为1.4 ?和1.5 ?。
3 数据模拟结果、分析和讨论
3.1 空配体GBP动力学中存在多种亚稳态
利用分子动力学模拟和数据分析,根据上下两区域的相对位置(两区域不同的相对位置表征为不同的开口角度和扭转角度),本研究确定了空配体GBP动力学中的5种亚稳定构象状态(见图3),并进一步根据绝对域间取向测量对开口构象和扭转构象进行了分类[19]。如图3所示,开放状态(状态S3,用蓝色表示)的占比最高(约64%)。笔者还观察了其他的亚稳态构象,包括扭转状态(S0状态,红色显示)、半扭转状态(S1状态,橙色显示)、半开放状态(S2状态,绿色显示)和逆时针扭转状态(S4状态,青色显示)。此外,发现这5种状态之间的平均转换时间非常快(< 300 ns)。
本模拟结果与核磁共振实验一致,表明GBP具有较高的构象灵活性,现有的晶体结构都不能很好地拟合残基偶极耦合(RDC)数据。此外,模型中稳定的开放状态(S3,占比>60%)与Talaga等之前的研究非常一致,在Talaga等的研究中观察到高达60%的高占比空配体开口构象状态[20]。然而,我们发现,无论是空配体闭口态还是空配体开口态,晶体结构似乎不位于任何局部最小值的MD模拟(见图3,粉色十字代表空配体闭口态晶体结构的相对角度,黑色十字代表配体闭口态晶体结构的相对角度)。
根据对晶体结构的研究,发现了两种典型的结构:空配体闭口结构(如1GCG[7])和空配体开口(如2FW0)。开口和闭口构象的共存被认为是将GBP与葡萄糖结合分解为“构象选择机制”的直接证据[5, 7-9, 21]。然而,没有观察到任何闭口的亚稳态构象,这表明在配体结合上达到配体闭口构象(即诱导拟合机制)需要显著的结构重排。另一方面,配体也可能选择与模型中确定的半开口构象结合的配体,进而诱导构象变化到结合态。结果表明,构象选择和诱导拟合都可能在GBP的结合机制中发挥作用。
3.2 蛋白结构域相互作用在确定空配体GBP动态方面起着至关重要的作用
场域静电相互作用对稳定识别的亚稳态构象起着至关重要的作用。如图4所示,空配体闭口晶体结构的两个域之间的静电结合自由能为>62 kJ·mol-1[崔兆宁5] 。这种强烈的排斥作用可能促使蛋白质打开。这种闭口构象的张开运动和扭转运动均可显著减小结构域间排斥,从而产生许多亚稳态构象。事实上,具有一定开度和扭转度的所有状态(S0-S4)都表现出相对较小的结构域间排斥相互作用(<25 kJ·mol-1[崔兆宁6] )。开放状态(S3)占主导地位,因为当蛋白以较大角度打开时,蛋白结构域相互作用几乎为0。有趣的是,排斥相互作用的大小與各态的平衡总体呈负相关,这证实了蛋白结构域相互作用在决定GBP构象动力学中起主导作用。进一步的研究表明,GBP结合界面上的大部分残基不是带电的就是亲水的,这排除了两个域之间像其他双域蛋白一样发生疏水靠近的可能性[15]。
此外,在实验中发现了一些降低与葡萄糖结合亲和力的GBP突变体,如E149C、E149C/ A213S/L238S等,可能会改变两个域之间的相互作用。在空配体闭口构象的两个域之间存在一个盐桥Lys92-Glu149 (K92-E149)。如果我们通过改变Lys92和Glu149侧链的方向来打破盐桥,那么相互作用的势能会受到显著的影响(从-39.7 kJ·mol-1到-18.4 kJ·mol-1[崔兆宁7] )。事实上,像E149C这样的突变也会改变相互作用电位(从-39.7 kJ·mol-1到-19.2 kJ·mol-1[崔兆宁8] )。我们认为盐桥的缺失使得有界结构更难保持,从而降低了与葡萄糖的结合亲和力,这也暗示了蛋白结构域相互作用的重要性。
3.3 晶体堆积效应稳定了GBP的空配体闭口构象
模型表明,开口构象主导大肠杆菌GBP蛋白的结构集合。然而,在相关种鼠伤寒沙门氏菌(PDB ID: 1GCG)中存在一个空配体闭口晶体结构。为了研究apo GBP在晶体中的稳定性,还建立了考虑晶体堆积效应的模拟。将GBP晶体结构放置在水盒的中心,并将20 ?内对称伴侣的所有残基都包含到中心GBP中。为了模拟结晶环境,在模拟过程中抑制了对称残基的重原子,分别获得了1GCG和2FW0的3×20ns轨迹。计算结果表明,无论是1GCG晶体还是2FW0晶体,GBP几乎不可能发生任何骨干运动。这一观察解释了为什么在晶体环境中比在溶液中更容易发现空配体闭口结构。
如以往许多光谱学研究中所讨论的,晶体堆积可能会影响开度或扭转角,从而使单晶结构难以代表溶液的状态。例如有研究表明,麦芽糖结合蛋白(MBP)的X射线晶体结构和核磁共振β-cyclodxtrin之间就存在开口角度10?的结构差异[22]。此外,MD模拟揭示的构象变化可能比晶体结构中揭示得更为明显和直接[23-24]。
3.4 影响铰链区域体积的突变体
对于一些PBPs,铰链残基庞大的侧链也会影响蛋白质动力学[25]。对铰链更小(L255A、N256A)和铰链更大(G109A/T110S)的突变体进行了控制模拟。随着铰区体积的减小,GBP仍有打开的趋势。这一结果表明,铰链残基侧链的粗大程度可能并不是引起开口运动的必要因素。Ortega等曾报道G109A/T110S双突变体在NMR实验中诱导GBP构象发生巨大转移。与他们的发现一致,双突变体的模拟结果表明,在铰链区域有更多的大体积残基可以产生更大的开口角的构象。综上所述,铰链残余侧链的大小对GBP的域间运动有一定的影响,但不是决定因素。
4 结 论
综上所述,利用分子动力学模拟和数据建模研究了空配体GBP的构象动力学。结果发现,场域静电相互作用在空配体GBP的构象动力学中起着至关重要的作用。通过马尔可夫模型(MSMs)分析确定了5种具有不同域方向的亚稳态,它们具有不同的结构域间静电排斥相互作用。在溶剂中,5种构象之间存在快速平衡(平均转化时间小于300 ns)。而铰链区域作为域间方向的互补调制器,施加了与蛋白结构域相互作用相平衡的几何约束。通过考虑蛋白结构域相互作用,成功地解释了一些现有的GBP突变体及其对其结合亲和力的影响。与晶体堆积效应有关的强排斥相互作用也解釋了溶液中X射线晶体结构与核磁共振结果之间的矛盾。
参考文献:
[1]BERNTSSON R P. A structural classification of substrate-binding proteins[J]. FEBS Lett, 2010,584(12): 2606-2617.
[2]SACKS D B. Guidelines and recommendations for laboratory analysis in the diagnosis and management of diabetes mellitus[J]. Clin Chem, 2002,48(3): 436-472.
[3]MARVIN J S,HELLINGA H W. Engineering biosensors by introducing fluorescent allosteric signal transducers: Construction of a novel glucose sensor[J]. Journal of the American Chemical Society, 1998,120(1): 7-11.
[4]AMISS T J. Engineering and rapid selection of a low-affinity glucose/galactose-binding protein for a glucose biosensor[J]. Protein Sci, 2007,16(11): 2350-2359.
[5]VYAS N K, VYAS M N,QUIOCHO F A. The 3 A resolution structure of a D-galactose-binding protein for transport and chemotaxis in Escherichia coli[J]. Proc Natl Acad Sci U S A, 1983,80(7): 1792-1796.
[6]VYAS N K, VYAS M N,QUIOCHO F A.Comparison of the periplasmic receptors for L-arabinose, D-glucose/D-galactose, and D-ribose. Structural and Functional Similarity[J]. J Biol Chem, 1991,266(8): 5226-5237.
[7]FLOCCO M M,MOWBRAY S L. The 1.9 Angstrom X-Ray Structure of a Closed Unliganded Form of the Periplasmic Glucose/Galactose Receptor from Salmonella-Typhimurium[J]. Journal of Biological Chemistry, 1994,269(12): 8931-8936.
[8]FLOCCO M M,MOWBRAY S L. Strange bedfellows: interactions between acidic side-chains in proteins[J]. J Mol Biol, 1995, 254 (1): 96-105.
[9]ORTEGA G. Carbohydrate Affinity for the Glucose-Galactose Binding Protein Is Regulated by Allosteric Domain Motions[J]. Journal of the American Chemical Society, 2012,134(48): 19869-19876.
[10]BORROK M J, KIESSLING L L,FOREST K T. Conformational changes of glucose/galactose-binding protein illuminated by open, unliganded, and ultra-high-resolution ligand-bound structures[J]. Protein Sci, 2007, 16(6): 1032-1041.
[11]HSIEH H V. Direct detection of glucose by surface plasmon resonance with bacterial glucose/galactose-binding protein[J]. Biosensors & Bioelectronics, 2004,19(7): 653-660.
[12]DER B S,DATTELBAUM J D. Construction of a reagentless glucose biosensor using molecular exciton luminescence[J]. Analytical Biochemistry, 2008,375(1):132-140.
[13]LEE Y H. Treponema pallidum TroA is a periplasmic zinc-binding protein with a helical backbone[J]. Nature Structural Biology,1999, 6(7): 628-633.
[14]SINHA N, KUMAR S,NUSSINOV R. Interdomain interactions in hinge-bending transitions[J]. Structure, 2001, 9(12): 1165-1181.
[15]ZHOU R H. Hydrophobic collapse in multidomain protein folding[J]. Science, 2004,305(5690): 1605-1609.
[16]WANG T. How optimal are the binding energetics of barnase and barstar [J].Biophysical Journal, 2004,87(3): 1618-1630.
[17]BAKER N A. Electrostatics of nanosystems: Application to microtubules and the ribosome[J]. Proc Natl Acad Sci U S A, 2001, 98(18): 10037-10041.
[18]BRUCCOLERI R E, NOVOTNY J,DAVIS M E. Finite difference Poisson-Boltzmann electrostatic calculations: Increased accuracy achieved by harmonic dielectric smoothing and charge antialiasing[J]. Journal of Computational Chemistry, 1997,18(2): 268-276.
[19]RAVINDRANATHAN K P, GALLICCHIO E,LEVY R M. Conformational equilibria and free energy profiles for the allosteric transition of the ribose-binding protein[J]. J Mol Biol, 2005,353(1): 196-210.
[20]MESSINA T C,TALAGA D S. Protein free energy landscapes remodeled by ligand binding[J]. Biophysical Journal, 2007, 93 (2): 579-585.
[21]VYAS N K, VYAS M N,QUIOCHO F A. Comparison of the Periplasmic Receptors for L-Arabinose, D-Glucose D-Galactose, and D-Ribose-Structural and Functional Similarity[J]. Journal of Biological Chemistry, 1991,266(8): 5226-5237.
[22]EVENAS J. Ligand-induced structural changes to maltodextrin- binding protein as studied by solution NMR spectroscopy[J]. J Mol Biol, 2001,309(4):961-974.
[23]KANDT C, XU Z T,TIELEMAN D P. Opening and closing motions in the periplasmic vitamin B-12 binding protein BtuF[J]. Biochemistry, 2006,45(44): 13284-13292.
[24]LIU M. Study on the mechanism of the BtuF periplasmic-binding protein for vitamin B-12[J]. Biophysical Chemistry, 2008,135(1-3): 19-24.
[25]ORTEGA G. Carbohydrate affinity for the glucose-galactose binding protein is regulated by allosteric domain motions[J]. J Am Chem Soc, 2012,134(48): 19869-19876.
收稿日期:2020-06-03
作者簡介:崔兆宁(1987-),男,中级工程师,博士,研究方向:高分子化学与化工。E-mail: cuizhaoning@cofco.com。
通讯作者:佟毅(1963-),男,正高级工程师,博士,研究方向:玉米深加工。E-mail: tongyi@cofco.com。
