光度法测定聚酯切片端羧基含量的方法条件优化
2020-08-05周丽华
周丽华,季 轩
(1. 中国石化仪征化纤有限责任公司研究院,江苏仪征 211900; 2. 江苏省高性能纤维重点实验室,江苏仪征 211900)
以PTA为原料的聚酯中,端羧基(-COOH)主要来源于未反应的对苯二甲酸残留羧基、酯键水解、酯键热降解、酯键热氧降解和端羟基热降解,而聚酯的端羧基含量是表征切片质量的重要指标,其含量的高低直接影响聚酯产品的品质,如聚酯热稳定性、纺丝质量、产品色相等。化学容量法测定聚酯端羧基含量具有操作方便、结果准确、分析成本低等优点,因而被广泛使用于聚酯行业,但由于聚酯端羧基的酸性较弱,需在非水相介质中方可进行测定,近年来随着改性聚酯研究的热增,特别是有色聚酯,目前使用化学容量法测定有色聚酯端羧基含量存在终点难以判断的问题,已无法满足现代分析的要求。而光度法在聚酯端羧基含量的测定中具有一定的优势,其原理是根据溶液颜色的变化,使用光度计将透光率转变为电信号并传递给滴定仪,滴定仪输出滴定曲线,自动判定滴定终点,解决了因人工无法判定终点和滴定误差的问题。
结合实际的分析情况,笔者对光度法测定聚酯端羧基含量的方法条件进行优化,为今后有色聚酯的端羧基含量测定提供分析支撑。
1 实验部分
1.1 仪器与设备
仪器与设备如表1所示。

表1 仪器与设备
1.2 原料及试剂
原料与试剂如表2所示。
表2 原料与试剂
1.3 测试原理
试样在混合溶剂中加热回流溶解,冷却后加入溴酚蓝作指示剂,溶液颜色的变化会导致透光率也随之改变,使用光度计把透光率转变为电信号并传递给滴定仪,滴定仪输出滴定曲线,同时自动判定滴定终点,并记录滴定终点所对应消耗滴定剂的体积,经计算后可精确得出试样中端羧基含量。
1.4 试验步骤
1.4.1 制样
根据滴定消耗的标准滴定溶液量不超过3 mL的原则[1],选定合适的实验室样品称样量,推荐样品的称样量如表3所示:

表3 不同端羧基含量所对应的称样量
1.4.2 KOH-C2H5OH标准滴定溶液(0.015 mol/L)
(1) 配制
量取150 mL按GB/T 601—2002[2]规定配制的浓度约为0.05 mol/L的KOH-C2H5OH标准滴定溶液,用乙醇稀释至500 mL,混匀。
(2) 标定
准确称取约0.035 g的基准试剂邻苯二甲酸氢钾(105~110 ℃干燥恒重)置于洁净烧杯中,加入50 mL蒸馏水完全溶解,用复合pH玻璃电极自动电位滴定仪标定其浓度。
1.4.3 样品测试
称取适量试样,精确至0.1 mg,放入磨口消解瓶中,加入50 mL混合溶剂,加热回流至试样完全溶解,冷却至室温。加入5~6滴溴酚蓝指示剂,将光度探头插入溶液液面1 cm以下,设置波长为600 nm,使用KOH-C2H5OH标准滴定溶液进行滴定,仪器自动判定滴定终点,记录标准滴定溶液消耗的体积,同一条件下进行空白试验。
2 光度电极测定端羧基的方法条件优化
2.1 溶液搅拌速度
在滴定过程中,待测溶液在磁力搅拌器中的搅拌速度对测定结果有着一定的影响,搅拌速度过小,标准滴定液与溶液混合不匀会影响反应速度;搅拌速度过大,会使溶液旋起产生大量气泡富集在光度电极的探头上影响光的透过率,进而给电极电位的稳定性带来较大的影响。试验中发现,根据选择搅拌磁子尺寸的不同,当旋起1/5溶液高的旋涡这样的搅拌速度[3]可以避免因旋起气泡多而对电极电位稳定性带来的影响。对纤维级聚酯切片样品在光度计探头有气泡和无气泡两种条件下进行6次重复性测定,结果见表4。

表4 搅拌速度引起气泡对测定结果的影响
从表4试验结果可见,富集在光度电极探头上的气泡给测试结果带来较大的影响,测试结果的RSD值较大,精密度差。
2.2 滴定模式的选择
溶剂空白的准确测定直接影响试样端羧基的结果,由于溶剂空白消耗KOH-C2H5OH标准溶液的体积较小,而且光度法测定空白溶液时,溴酚蓝指示剂由黄色变成蓝色的突变要早于人工判定的滴定终点,因此如何利用光度法准确测定空白尤其重要。在电位滴定仪的动态和静态两种滴定模式下,进行6次空白的重复性测定,结果见表5,同时在两种模式下对不同端羧基含量的聚酯切片进行6次分析测定,取其平均值,结果见表6。
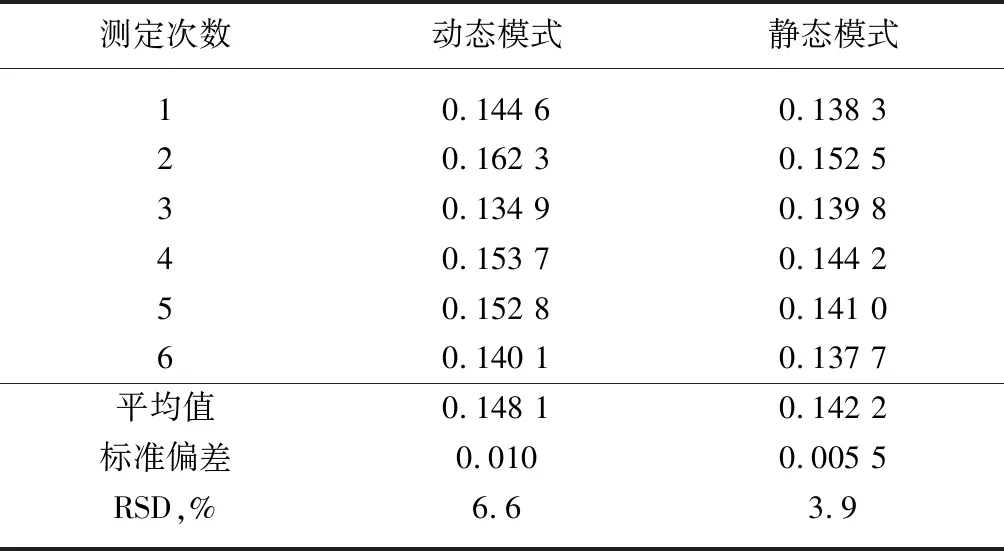
表5 动态模式和静态模式下测定的溶剂空白值 单位:mL
动态模式,加液的步长是滴定仪根据信号变化计算出来的。
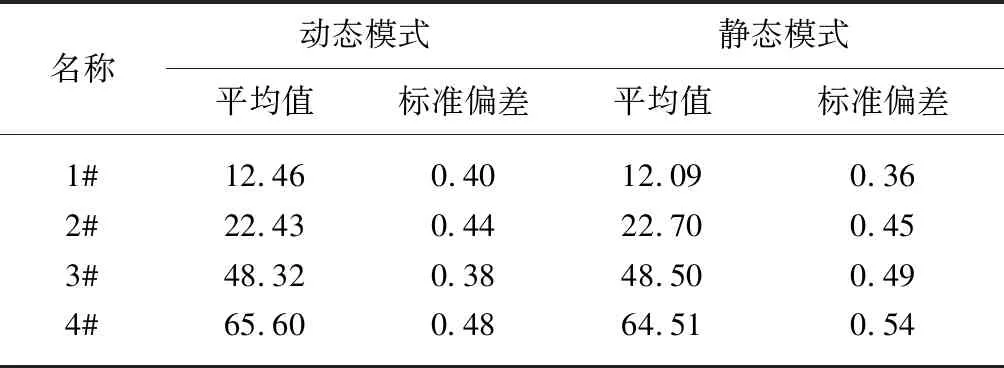
表6 动态模式和静态模式下测定聚酯切片的端羧基 单位:mol/t
从表5试验结果可见,两种模式下测得溶剂空白值存在着一定的差异,动态模式下测得空白值的RSD值略大于静态模式的结果,静态模式下测得空白值的标准偏差相对较小,精密度较好;从表6试验结果可见,两种模式下测得聚酯端羧基结果的差异不大,重复性好。因此光度法测定溶剂空白优选在电位滴定仪的静态滴定模式下进行,而样品测试选择动态滴定模式,缩短了分析时间,提高了工作效率。
2.3 混合溶剂的选择
由于邻甲酚的极性强,用于聚酯端羧基测试效果优于使用苯酚,在ASTM D7409—2007[4]、FZ/T 50012—2006和GB/T 14190—2017[5]标准中的光度法均使用邻甲酚/三氯甲烷混合溶剂,但是邻甲酚易氧化,在实验中发现邻甲酚经蒸馏后使用效果较好。考虑到配制邻甲酚混合溶剂的前处理操作及其对试验人员的危害性,笔者在使用苯酚/三氯甲烷和邻甲酚/三氯甲烷作为消解液下,对纤维级聚酯标准切片的端羧基含量进行6次分析测定,容量法的标准值为(21.50±1.80)mol/t,结果见表7。
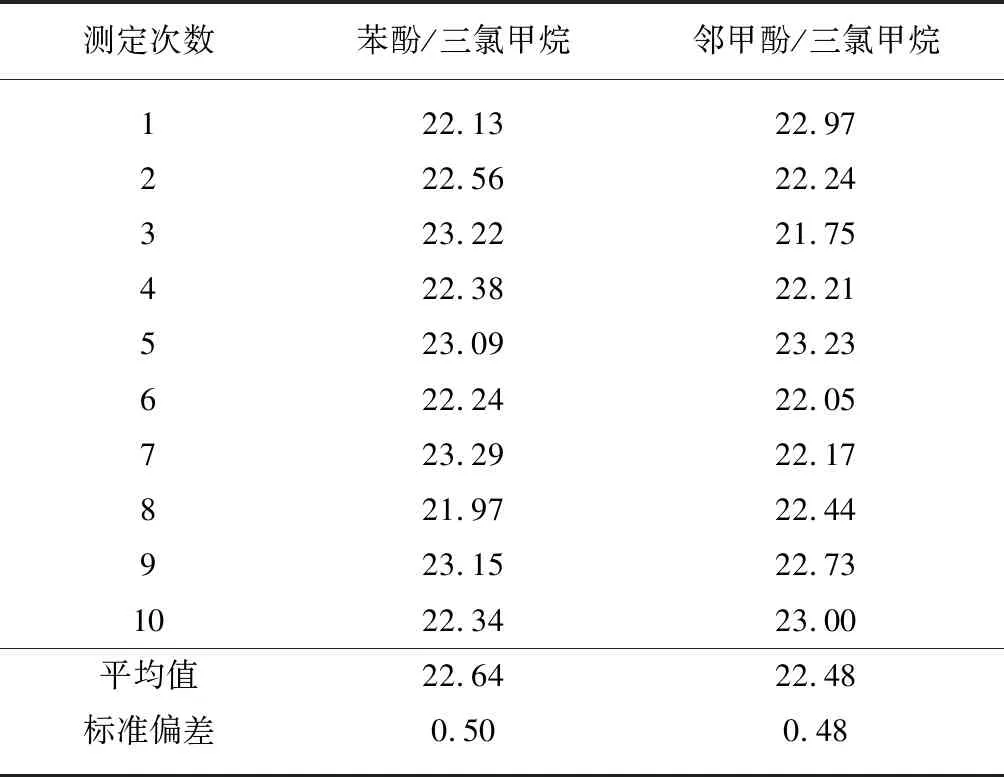
表7 不同消解液下测定纤维级聚酯切片的端羧基含量 单位:mol/t
从表7试验结果可见,使用两种不同的消解液,端羧基含量的测试结果差异不大,且从平均值上可以看出光度法测得结果均高于标准值的中心值,但结果均在标准值范围内,而且标准偏差小于1%,重复性较好。邻甲酚相对于苯酚来说其危害性比苯酚强些,因此从绿色环保和安全的角度考虑,选择毒性相对较低的苯酚溶剂亦能够满足聚酯端羧基含量的测定工作。
2.4 标准滴定液的稳定性
试验中使用KOH-C2H5OH标准滴定溶液的浓度偏低,大大减小了因消耗滴定体积过小而引起的分析误差,但在实际操作中,随着存放时间的延长,KOH-C2H5OH标准滴定溶液会吸收空气中的二氧化碳使得浓度发生变化,直接影响端羧基测定结果的准确性。为了考察标准滴定液的稳定性,在不同存放时间下对标准滴定溶液的浓度进行标定,试验如结果表8所示,5天内标准滴定液的浓度相对较稳定,5天以后标准液的浓度逐渐变小,影响测试结果的准确性。
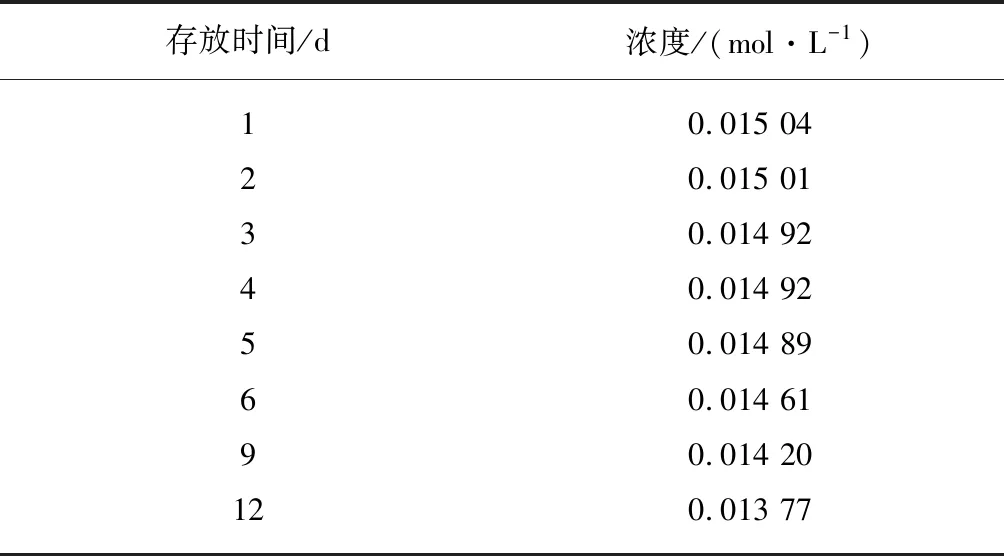
表8 存放时间对KOH-C2H5OH标准滴定溶液浓度变化的影响
2.5 光度法测定聚酯端羧基结果准确度的考察
为了考察光度法测定聚酯端羧基结果的准确性,在不同实验室使用苯酚/三氯甲烷混合溶剂测定4个工业丝样品的端羧基含量,每个样品取其平均值,测试结果如表9,同时对纤维级聚酯标准切片和瓶级聚酯切片分别进行10次重复性试验,纤维级容量法的标准值为(21.50±1.80)mol/t,瓶级容量法的标准值(16.76±1.17)mol/t,测试结果如表10。
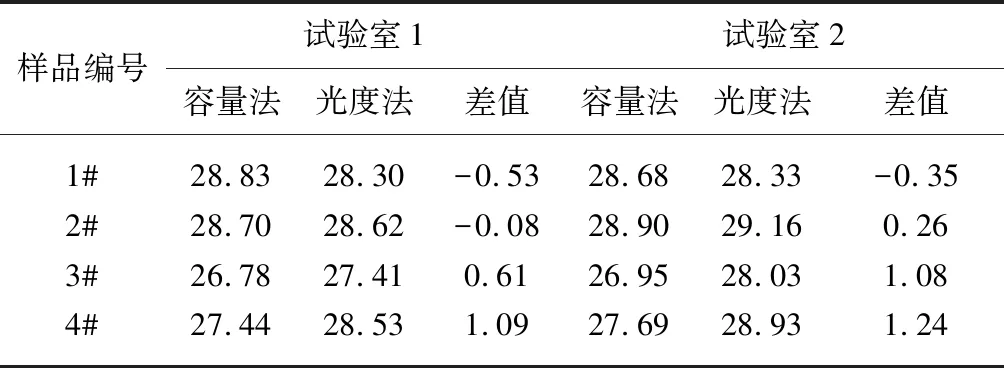
表9 不同试验室间样品的端羧基测定结果 单位:mol/t

表10 聚酯标准切片的端羧基测定结果 单位:mol/t
从表9试验结果可见,试验室1中光度法与容量法分析结果的差值为(-0.53~1.09)mol/t,试验室2中光度法与容量法分析结果的差值为(-0.35~1.24)mol/t,均在合理误差范围内。从表10试验结果可见,与容量法的标准值相比较,光度法测定聚酯标准切片的端羧基结果较高些,但均在标准值的范围内,测试结果的标准偏差0.50,重复性较好。光度法测定聚酯端羧基结果准确可靠,满足工业生产的应用。
3 有色聚酯端羧基的测定
随着改性差别化、功能性聚酯研究的热增,尤其是添加有色基团的改性聚酯,由于人工判断滴定终点误差较大,容量法已无法满足改性聚酯端羧基含量的测定。实验室使用光度法测定红、蓝、灰和黑四种聚酯切片的端羧基含量,每个样品测定6次,试验结果如表11。
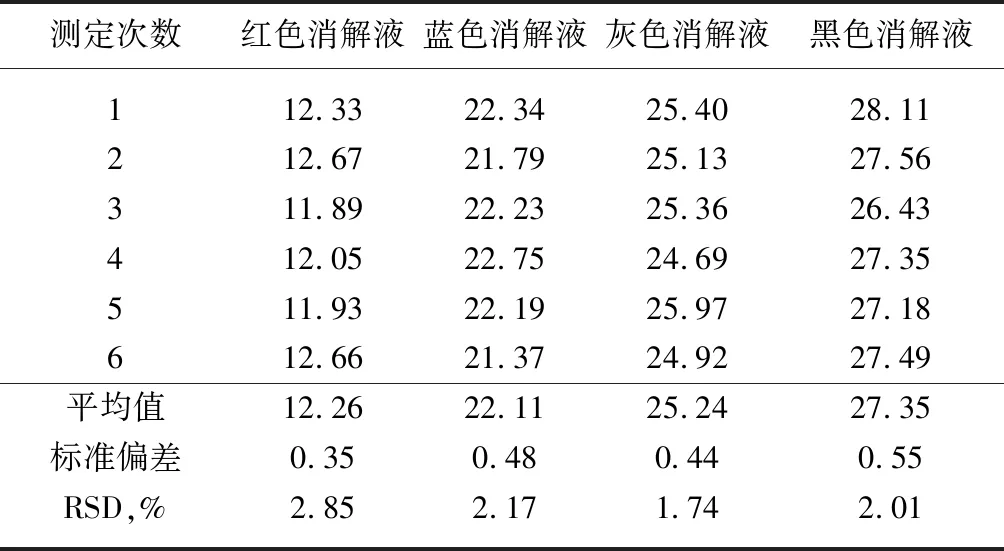
表11 光度法测定有色聚酯样品的端羧基含量 单位:mol/t
根据表11可见,在对红、蓝、灰和黑四种不同颜色聚酯消解液的分析中,光度法测定样品端羧基结果的RSD值小于3%,精密度好,解决了因容量法无法判定终点所带来的困扰。
4 结 论
在光度法测定聚酯端羧基含量的分析过程中,通过对溶液搅拌速度、空白溶剂测定、混合溶剂的选择、标准滴定液的稳定性以及不同实验室间的试验分析发现,选择苯酚溶剂亦能够满足测试,优选在电位滴定仪的静态滴定模式下准确测定空白溶剂,标准滴定液的浓度在5 d内相对较稳定,光度法测定聚酯切片端羧基结果的准确度较好,解决了容量法无法测定有色聚酯端羧基的困扰,避免容量法因人为因素导致的分析误差。
