基于分组贝叶斯排序的药物-靶标关系预测
2020-08-03丁棋梁石泽智李建华
丁棋梁,石泽智,李建华
华东理工大学 信息科学与工程学院,上海 200237
1 引言
计算机辅助药物设计是一个跨学科的研究领域,包括对生物学、化学、物理学和信息学的研究,其目的是加速药物研发过程。药物研发的关键是寻找药物和靶标间是否存在相互作用关系(Drug-Target Interaction,DTI)。尽管可以通过体内外测定[1]药物和靶标间是否存在相互作用,但这些方法时间长且成本昂贵[2]。因此,可以利用计算机技术来预测可能的DTI,通过实验来筛选药物[3],可以显著降低向市场推出新药的成本[4]。
目前主要有两类计算机预测DTI方法:对接模拟和机器学习方法。对接模拟方法[5]是利用靶标的3D结构来鉴定与药物是否存在潜在结合位点,但是非常耗时且需要靶标的3D结构,并不是所有的靶标都具有3D结构。一些研究人员报告,标准的分子对接评分函数可能被基于机器学习的评分函数所取代,并具有改进的预测结果[6]。机器学习方法通常利用药物和靶标结构的特征[7]、药物的副作用[8]以及已经确认的DTI的知识[9]。
近年来,机器学习技术的迅速发展为预测DTI提供了有效的方法,基于机器学习的方法大致分为四类:分类方法、矩阵分解方法、核方法和网络推理方法。支持向量机(SVM)是一种经典的分类方法,目前已经被Nagamine[10]和Wang等人[11]使用来预测DTI。矩阵分解的两种代表性方法是双核的核化贝叶斯矩阵分解(KBMF2K)[12]和多相似协同矩阵分解(MSCMF)[13]。核方法主要包括药物-靶标对核方法(PKM)[14]、网络拉普拉斯正则化最小二乘法(NetLapRLS)[15]和具有Kromecker积核的正则化最小二乘法(RLS-Kron)[16]。Bleakley和Yamanishi[17]建立了二部局部模型(BLM),并对药物-靶标相互作用网络进行学习,是一种典型的网络推理方法。然而,这些基本方法都没有预测新药物或新靶标的能力。为了预测新药物或新靶标,Mei等人[18]和Laarhoven等人[19]通过交互邻居信息解决了这个问题。
前面提到的方法侧重于预测所有未知药物-靶标对是否存在相互作用的概率,导致时间复杂度较高。为了降低时间复杂度,Ladislav等人[20]提出了一种新的思路,以药物为中心进行研究,分别对特定药物存在相互作用的靶标进行排序。靶标排名越靠前就最有可能与该药物存在相互作用,并根据预测的相互作用概率分别为每个药物确定未知靶标。他们使用贝叶斯个性化排序的矩阵分解技术(BPR-MF)来预测DTI,称为贝叶斯排序方法(Bayesian Ranking,BR)。虽然BR取得了很好的效果,但该方法的局限是所有药物间是相互独立的,无法使一些相似的药物产生互动。根据与特定靶标存在相互作用的药物间是存在相似性的现实,为了使这些相似的药物间产生互动,本文对这些相似药物进行了分组,并推导出分组贝叶斯排序的理论模型。最后通过实验验证,提高了其性能。
2 原理及相关工作
在这一部分,首先描述了在研究中使用的数据集,以及获得药物和靶标相似度矩阵的过程。然后详细描述了药物-靶标预测问题,并在DTI预测的背景下引入贝叶斯排序方法。最后详细描述采用贝叶斯排序方法对药物-靶标关系预测的优势。
2.1 原理
本文使用了五个公开的药物-靶标相互作用数据集,即核受体(NR)、G蛋白偶联受体(GPCR)、离子通道(IC)、酶(E)和激酶(Kinase)。表1给出了每个数据集的一些统计数据,包括药物总数、靶标总数、已知相互作用总数和最近验证的相互作用总数。

表1 数据集统计
每个数据集包含三个矩阵:(1)药物-靶标相互作用矩阵;(2)药物相似度矩阵;(3)靶标相似度矩阵。一般来说,药物相似度和靶标相似度可以用多种方法计算。本文采用与对比方法同样的算法来计算靶标相似度和药物相似度,通过序列对齐方法计算靶标相似度,如Smith-Waterman算法。除Kinase数据集外,其余四个数据集都通过SIMCOMP[21]方法计算药物相似度;而在Kinase数据集中,则通过2D Tanimoto系数计算药物相似度。
2.2 基本符号及问题描述
在本文中,假设有m个药物和n个靶标,D是所有药物的集合,T是所有靶标的集合。药物与靶标之间的相互作用关系用二元矩阵Y∈Rm×n表示,其中每个元素yij∈{0,1}。如果药物已经被实验验证与靶标存在相互作用,则设为1;否则,设为0。定义新药物集和新靶标集合TN=药物相似度矩阵用SD∈Rm×m表示,靶标相似度矩阵用n×n表示。
矩阵分解法的目的是将药物和靶标映射到一个共享的潜在空间,其中f表示其维数(潜在因子的个数),ui∈Rf表示药物di的潜在因子,表示靶标tj的潜在因子。定义U∈Rm×f为所有药物潜在因子的矩阵,V∈Rn×f为所有靶标潜在因子的矩阵。药物di与靶标tj相互作用的预测概率r̂ij定义为其潜在因子的点积,因此最终预测药物与靶标之间的相互作用关系矩阵Ŷ可以用Ŷ=UVT来表示。进一步将每种药物的训练集定义为三元组训练集Ds⊂D×
本文以药物为中心的重定位方法预测DTI。主要目标是对任意药物d∈D,提供所有靶标的排序,排名最靠前的靶标以最大的概率与药物d产生相互作用。
2.3 贝叶斯排序方法
贝叶斯排序方法是建立在BPR-MF算法三大假设上,而BPR-MF算法参考了文献[20]。如下是BPR-MF算法基于的三大假设:
(1)药物和靶标之间的相互作用行为是彼此独立的。
(2)药物和靶标的特征矩阵均服从高斯分布,且平均值为0,方差为常数。
(3)药物-靶标相互作用关系矩阵的预测值和真实值之间的误差需要满足均值为0,方差为常数的高斯分布。
本文采用的是贝叶斯排序和矩阵分解的组合方法,并记为BPR-MF,它是建立在3个基础假设上的。首先基于这些假设建立相应的概率模型,然后利用贝叶斯公式,最大化后验概率,建立起对应的优化准则,最后对其进行求解获得相应的药物和靶标特征矩阵,进而重构药物-靶标关系网络进行未知药物-靶标关系的预测。
为了使每个药物尽可能找到其所有正确的靶标排序,需要用贝叶斯公式最大化如下后验概率:
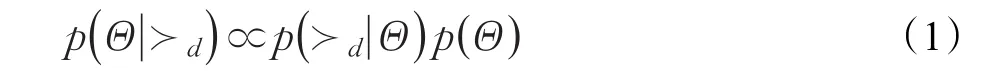
其中,Θ表示矩阵分解参数。基于假设(1)可以得到特定药物的概率函数p(≻d|Θ)用以下公式来表示:

药物d与靶标tj相互作用的概率大于该药物与靶标tk相互作用的概率定义如下:
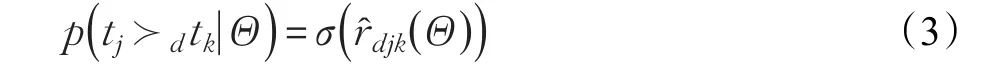

2.4 贝叶斯排序方法的优势
贝叶斯排序方法的一个核心步骤是构建新的训练集,不同的是,这里的训练样本不是药物-靶标对,而是一个由药物和靶标组成的三元组,这里记为(d,ti,tj),其中药物d与靶标ti存在相互作用关系,而与靶标tj的相互作用关系未知。
贝叶斯排序方法使用三元组作为新的训练集,与传统方法进行对比,不再需要对所有未知药物-靶标对是否存在相互作用关系进行预测,只需要对特定药物存在相互作用的靶标进行排序。靶标排名越靠前就最有可能与该药物存在相互作用,并根据预测的相互作用概率分别为每个药物确定未知靶标,可以明显降低时间复杂度。
3 分组贝叶斯排序方法
在这一部分,首先描述两个新定义,然后提出新假设及成立的依据。最后在新假设的基础上推导出分组贝叶斯排序(Group Bayesian Ranking,GBR)的理论模型,来平滑新药物和新靶标。
3.1 分组思想介绍
定义1(个体相互作用)个体相互作用是药物di和靶标tj之间相互作用的概率。例如,药物di和靶标tj之间相互作用的概率表示为
定义2(分组相互作用)分组相互作用是对特定靶标存在相互作用的药物集合和该靶标存在相互作用的概率。例如,药物集合G和靶标tj之间相互作用的概率表示为。其中表示已知与靶标tj存在相互作用的全体药物集合。
本文在分析本地和远程生物信息系统模型基础上,提出了一个基于数据仓库的架构思想的、适用于病毒序列数据库的集成系统架构。其目的是实现对病毒序列数据的分类提炼、整理和系统化,并提供相应的集成分析服务。同时以流感病毒序列为例,建立了一个流感病毒序列集成数据库系统,为相关数据库的构建积累了一定的经验。下一步将对更多病毒类别(如肠道病毒、腺病毒等)的数据进行集成,进一步扩充和完善现有的病毒序列集成数据库系统。
新假设:如果药物-靶标对(di,tj)是已知存在相互作用关系的,而药物-靶标对(di,tk)是否存在相互作用是未知的,本文提出的新假设用以下式子进行表示:

其中,且。通过图1可以更加直观地推出新假设。药物d1,d2,d3与靶标t1是已知存在相互作用的,而药物d1与靶标t2是否存在相互作用是未知的。根据定义 1,都大于r̂,所以12r̂12也成立,即,得到新假设:(G,t1)≻(d1,t2),这里取G={d1,d2,d3}。

图1 药物-靶标相互作用关系图
分组贝叶斯排序方法具体实现步骤如算法1所示。
算法1分组贝叶斯排序方法
输入:相互作用矩阵Y;相似性矩阵SD,ST;药物(或靶标)邻居k的大小。
输出:更新的相互作用矩阵Ŷ。
步骤1初始化U,V,b。
步骤2将SD,ST更改为仅包含每个项的前k个最近邻居。
步骤3使每个药物靶标对(di,tj),满足rij=1。
步骤4随机选择靶标tk,使rik=0。
步骤5对特定靶标tj存在相互作用的药物进行随机分组,使分组大小|G|=1,2,3,4,5。
步骤6 更新bj,bk,ui,vj,vk。
步骤7返回步骤3,直到达到最大迭代次数。
3.2 新假设成立依据
本文根据以下两个方面信息做出合理的假设:
(1)对于靶标:如果药物di和靶标tj存在相互作用,且能找到其他药物对靶标tj也存在相互作用,则药物di与靶标tj存在相互作用概率大于与靶标tk存在相互作用概率。所以可以用(G,tj)≻(di,tk)代替(di,tj)≻(di,tk)。
(2)对于药物:在所有对特定靶标tj存在相互作用的药物间引入互动是很自然的,因为意味着这些药物是存在相似关系的。有共同相似关系的药物组G⊆Dtrtj,它们对靶标tj都存在相互作用关系。
3.3 理论模型
为了更加精确地研究个体相互作用和分组相互作用对预测结果的不同程度影响,把它们线性地结合起来:
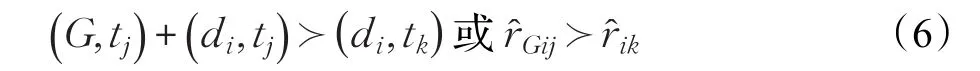
其中,r̂Gij=ρr̂Gj+(1-ρ)r̂ij。 0≤ρ≤1是用于融合两种不同相互作用的权衡参数,可通过测试验证集来确定。
有了上述假设,在BR基础上,用r̂Gij代替r̂ij,每个药物就有了新的靶标排序,称为分组贝叶斯排序。因此,最终分组贝叶斯排序方法目标函数如下:
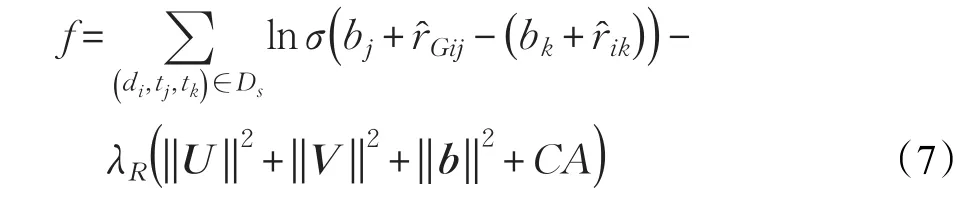
其中,bj和bk分别为靶标tj和靶标tk的偏差,b是所有靶标的偏差,CA是潜在因素距离的正则化项。设训练集中的一个三元组(di,tj,tk)∈Ds,CA可以用以下公式来表示:

本文使用广泛的随机梯度下降(SGD)优化目标函数f,模型参数Θ包括ui,vj,vk,bj和bk。首先需要计算目标函数中参数的梯度,然后根据相应的梯度,模型参数可以更新如下:

3.4 平滑新药物和新靶标
通过合并邻居信息进行预测新药物或新靶标的相互作用。显然,贝叶斯排序方法不能预测新药物和新靶标,只能通过负例(未知DTI)了解其潜在因素,这将严重破坏整个模型。因此,基于协同过滤的思想[22],本文整合了邻居信息,得到未知药物或未知靶标的潜在因素,如下所示:

其中,N+(di)和N+(tj)分别是已知药物和靶标k个最近邻居的集合。在实验中,k通常设置为5以简化模型。
4 实验及结果分析
4.1 评价指标
本文采用ROC曲线以下面积(Area Under the ROC Curve,AUC)、标准化折扣累计收益[8](normalized Discounted Cumulative Gain,nDCG)和平均精度(Mean Average Precision,MAP)作为评价指标。AUC值和MAP值几乎在所有药物-靶标关系预测中都用来做评价指标,而nDCG值是在最近文献[8]才提出的评价指标,具有很大的参考价值,所以本文将它纳入评价指标。
nDCG通过使用分级相关特征,能区分出具有较高潜在影响的DTI预测。nDCG通过自然截断仅考虑排名前k个对象对DTI预测所产生的影响,忽略了不重要对象所产生的较小影响,降低了时间复杂度。
4.2 实验设置及对比方法
为了与先前的研究方法[15,18,20]具有对比性,本文采取了5次10折交叉验证(CV)的实验评估了GBR预测方法的性能。并将该方法与5个典型的DTI预测方法进行了比较,如基于高斯核的加权最近邻[11](WNN-GIP)、协同矩阵分解[13](CMF)、网络拉普拉斯正则化最小二乘法[15](NetLapRLS)、最近邻居信息的二部局部模型[18](BLM-NII)和贝叶斯排序方法[20](BR)。
计算每一次交叉验证的平均值,并重复地运行5次,每次随机把已知DTI分成10份,得到一个最终的AUC值。并采用同样的方法计算出nDCG值和MAP值。
4.3 参数设置
在GBR方法中,参数对优化结果起着重要作用。从理论上不难发现,选取的邻居个数k越多,性能越好,但随着邻居个数不断增加到某个值时,性能改善并不明显,而时间复杂度将持续提高,导致算法效率低下。在药物-靶标关系预测中,随着分组大小||G值增加,性能会越好,但时间复杂度呈现指数级增加。当||G=1时,改进的方法就是BR方法了,所以选取合适的k值和||G值至关重要。
为了深入理解选取邻居个数和分组大小对GBR方法的影响,将参数调整范围设置为k∈{3,5,8,15,20,30}和|G|∈{1,2,3,4,5},通过实验来选取合适的k值和|G|值。从图2可以看出,当邻居个数k值大于8时,性能改善并不明显。从图3可以看出,当分组大小|G|值大于3时,nDCG改善明显减低,有些甚至下降。

图2 邻居个数的影响
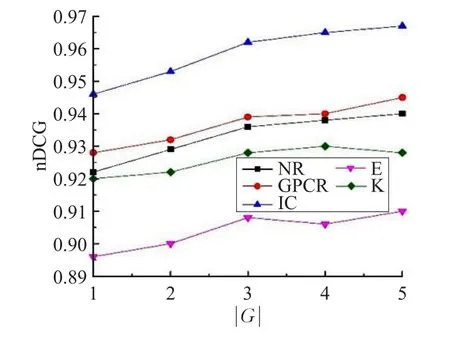
图3 分组大小的影响
在这个实验中,将最近邻的大小设置为k=8,分组大小设置为|G=3|。
4.4 与典型的五种方法进行比较
为了说明本文的GBR方法优于五种典型的DTI预测方法,本文与五种典型方法使用相同公开的数据集,相同的实验环境。结果汇总在表2~4中,正如所希望的:GBR方法在AUC、nDCG和MAP方面均优于典型方法。

表2 与典型算法的AUC值比较 %

表3 与典型算法的nDCG值比较 %

表4 与典型算法的MAP值比较 %
4.5 预测新的相互作用
这五个公开数据集是几年前提取的,并一直保持不变,以便于不同方法的比较。然而,这些数据集中一些未知的药物-靶标对,最近通过生物化学方法已被确认其相互作用,并已上传到数据库,如DrugBank[15]、KEGG[23]或Matador[24]。
本文的目的是证明GBR方法比典型方法更能准确地预测最近验证的药物-靶标对。若在当前数据库KEGG、DrugBank或Matador中含有预测的新相互作用,则预测成功。
为了证明GBR方法能够预测新的相互作用,本文使用了原始数据集的所有相互作用药物-靶标对,对GBR方法及典型方法进行了训练,并对非相互作用药物-靶标对进行了排序。本文只考虑了GPCR和Enzyme数据集中每种药物的前10个药物-靶标对。
图4显示了在GPCR数据集中的命中数,图5显示了在Enzyme数据集中的命中数。正如所期望的,在GPCR数据集中,GBR方法的前10个命中数占总数的63%;在Enzyme数据集中,GBR方法的前10个命中数占总数的34%,明显高于典型方法。
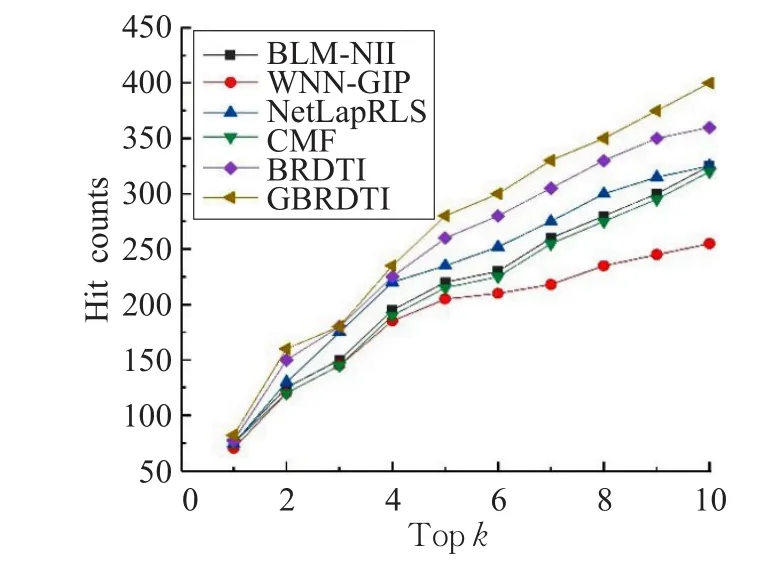
图4 GPCR数据集上的命中数

图5 Enzyme数据集上的命中数
最后,表5展示了多种药物-靶标预测方法在5个数据集上topN的(N=10,30)药物-靶标关系中预测成功的比例,所有的方法均使用优化后的参数进行实验。可以看到,GBR方法在所有数据集上的预测准确性都提高了。从横向看,GBR方法取得了8个最大值,而其他方法最好的情况也才取得3个最大值。
5 结语
本文考虑了分组相互作用对贝叶斯排序方法的影响,根据与特定靶标存在相互作用的药物具有相似性的现实,把这些相似的药物进行分组,得到分组的药物集合。然后根据分组的药物集合提出新假设,在新假设的基础上推导出分组贝叶斯排序的理论模型。最后,本文还结合了邻居信息来平滑新药物和新靶标的预测。通过相应的实验,证明了本文方法在性能上优于典型方法。为了将来的工作,计划开发一种新的对相似靶标进行分组方法以进一步改进性能。

表5 topN个药物-靶标关系中预测成功的比例
