丝氨酸手性分子与离子单重低激发态特性的理论计算
2020-07-18王冰杰
王 冰 杰
(白城师范学院 数学与统计学院, 吉林 白城 137000)
丝氨酸(Ser)是一种天然氨基酸, 是组成人体蛋白质必需的氨基酸之一, 具有较强的生物活性. 文献[1]采用高效液相色谱法, 在色谱柱Chiralpak AD-H手性柱上成功拆分了DL-苏式-对甲砜基苯丝氨酸乙酯; 文献[2]利用Ser分子的手性结构, 研究了桥联环糊精在手性识别中的应用; 文献[3]通过改进的合成方法制备了含Ser残基碱基为腺嘌呤的手性肽核酸单体; 文献[4]在碱性条件下将L-Ser和L-苏氨酸与氯乙醇发生反应, 分别合成了具有手性的螯合中心配体L-N,N-双(2-烃乙基)Ser和L-N,N-双(2-烃乙基)苏氨酸; 文献[5]实现了未衍生化D,L-Ser对映体的基线分离和检测; 文献[6]在优化的色谱条件下, 将Ser对映体进行拆分; 文献[7]以L-Ser为原料合成了NBoc-3-碘代-L-丙氨酸甲酯, 合成产率较高, 可为合成氟代酪氨酸提供中间体原料; 文献[8]基于L-Ser合成了具有旋光活性及环状磷脂的替加氟衍生物等.
电场作用下S型Ser(S-Ser)手性分子及离子单重低激发态特性的理论研究目前文献报道较少. 基于此, 本文利用电场作用模拟外界条件变化对手性S-Ser分子及负二价离子轨道跃迁等单重低激发态特性进行理论研究.
1 理论和计算方法
基于密度泛函理论(DFT)中的B3P86方法, 在6-311++G(d,p)基组水平上优化手性S-Ser分子、 正负一价离子及负二价离子的基态稳定几何构型, 并用含时密度泛函理论(TD-DFT)方法, 计算手性S-Ser分子及负二价离子轨道跃迁等单重低激发态特性. 所有计算均在Gaussian 09软件包内完成, 图形由GaussView5.0生成.
2 结果与讨论
2.1 手性S-Ser分子和离子的基态结构特性
2.1.1 手性S-Ser分子和离子的基态几何结构 对手性S-Ser分子的基态几何结构优化得到其稳定构型, 其能量数值等参数与文献[9]结果相符. 手性S-Ser分子、 正负一价离子及负二价离子的基态稳定构型如图1所示.

图1 S-Ser,S-Ser+,S-Ser-和S-Ser2-的结构
2.1.2 手性S-Ser分子和离子的结构参数 手性S-Ser分子和离子的键长(R)、 键角(A)和二面角(D)列于表1. 由表1可见: 键长R(1,5)随手性分子俘获电子数目的增加先增加后减小,R(1,4)和R(1,8)随手性分子俘获电子数目的增加先减小后增加; 键角A(1,8,10)和A(4,1,8)随手性分子俘获电子数目的增加而增加; 二面角D(9,8,1,12)随手性分子俘获电子数目的增加而减小; 手性S-Ser分子结构在剥离一个电子后发生明显变化, 出现断键现象.
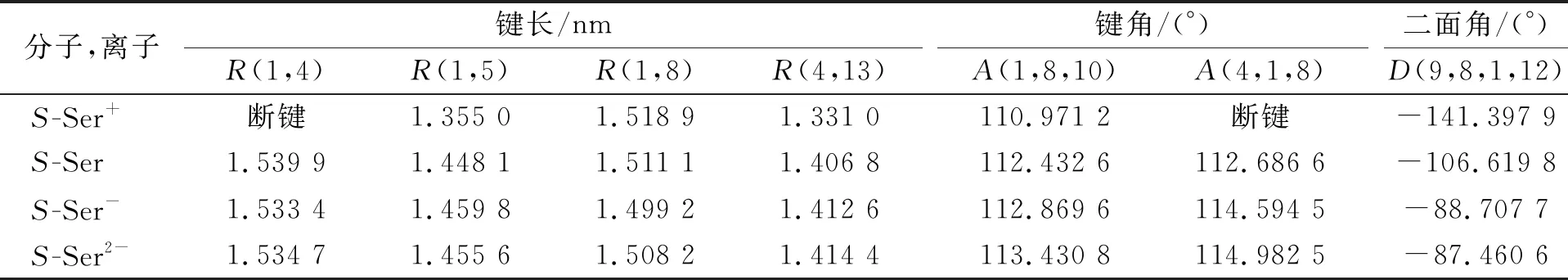
表1 手性S-Ser分子和离子的键长(R)、 键角(A)和二面角(D)
手性S-Ser分子和离子的电荷分布列于表2. 由表2可见: 1C和4C原子电荷分布随手性分子俘获电子数目的增加而增大, 5N,10O,13O,11H和12H原子电荷分布随手性分子俘获电子数目的增加而减小, 其中1C,4C,5N和12H原子电荷数值变化较大; 8C原子电荷分布随手性分子俘获电子数目的增加先减小后增加; 14H原子电荷分布随手性分子俘获电子数目的增加先增加后减小.

表2 手性S-Ser分子和离子的电荷分布
2.2 手性S-Ser分子单重低激发态特性
2.2.1 手性S-Ser分子轨道跃迁的单重低激发态特性 基于TD-DFT计算获得手性S-Ser分子各单重低激发态S1~S10的电子跃迁轨道, 结果列于表3.
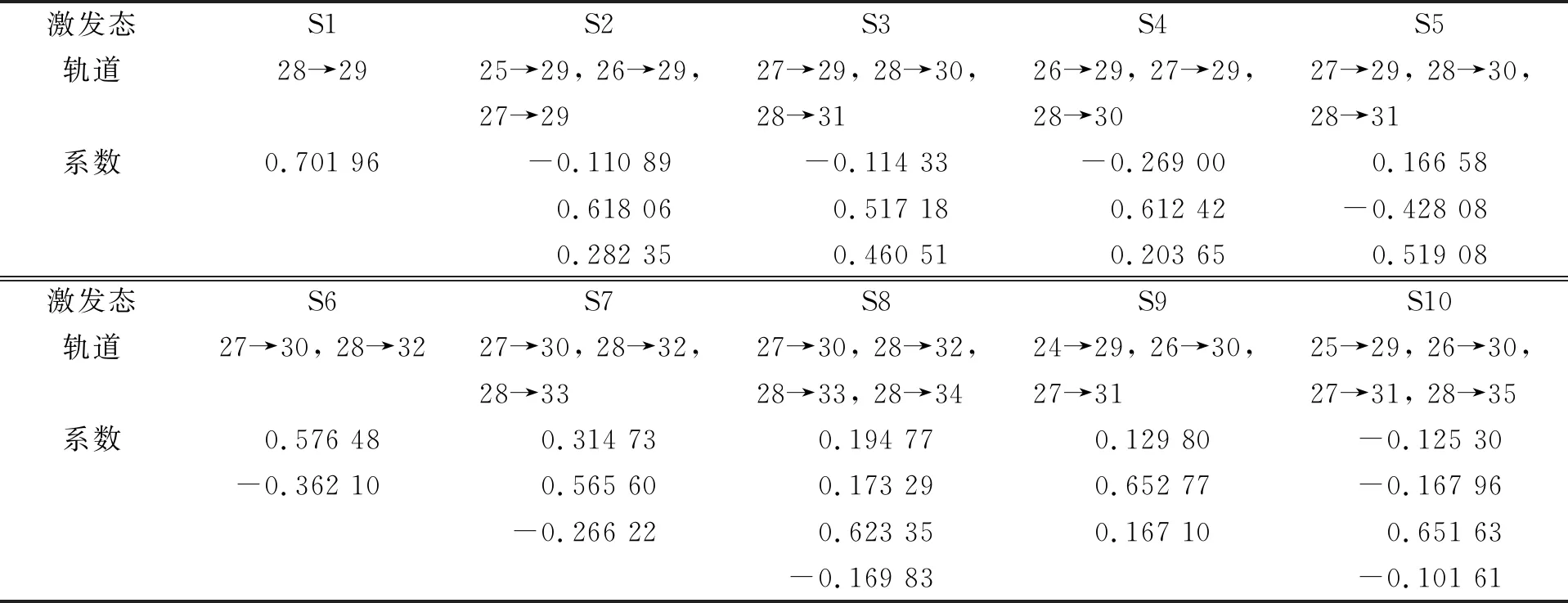
表3 手性S-Ser分子各激发态电子跃迁轨道
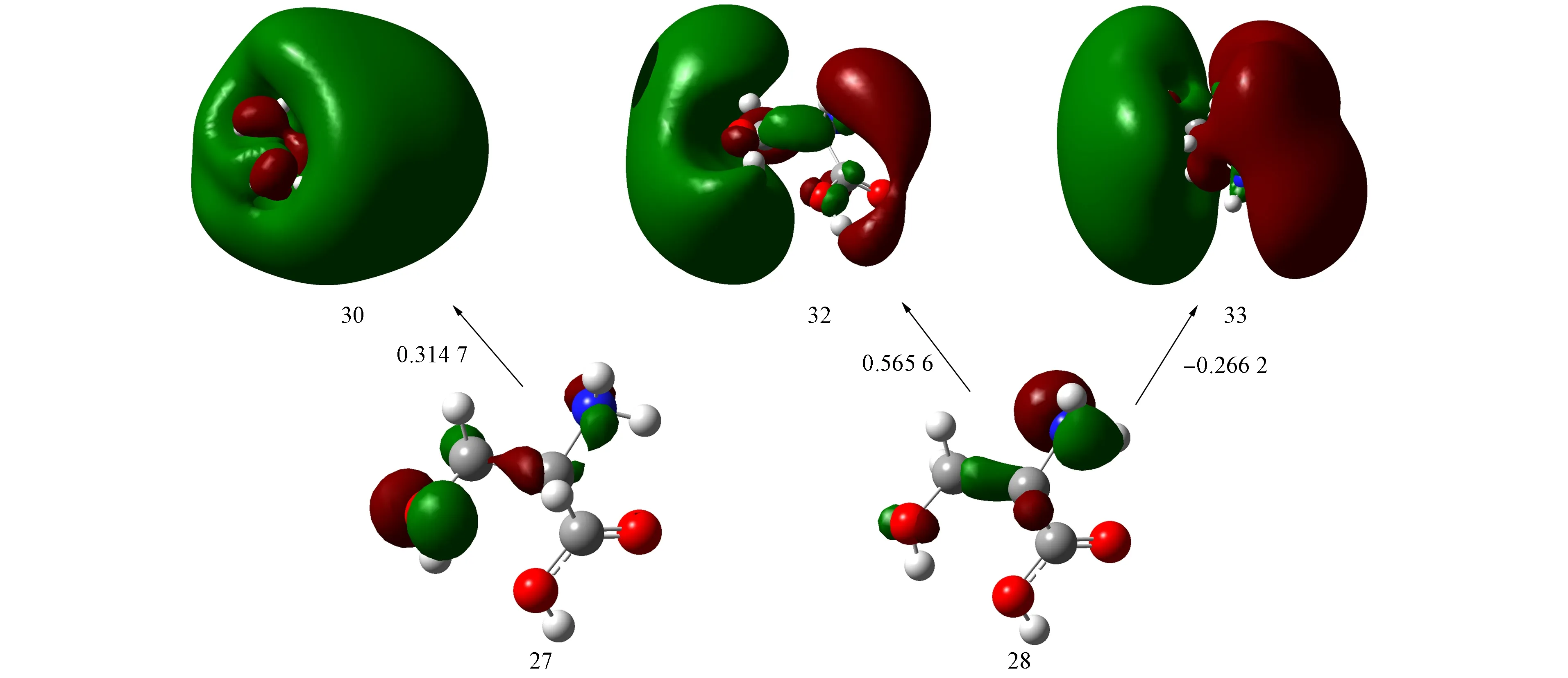
图2 手性S-Ser分子的轨道跃迁
以S7激发态为例, 标注了手性S-Ser分子轨道、 跃迁及对应系数, 如图2所示. 由图2可见: 轨道27为O13原子的孤对电子轨道, N5原子的孤对电子对C1—C4和C4—H2的σ键略有贡献, 可考虑为次要特征; 轨道28为N5原子的孤对电子轨道, O13原子的孤对电子对C1—C4和C1—C8的σ键略有贡献, 但可忽略不计; 轨道30为s型Rydberg轨道特征; 轨道32为p型Rydberg轨道特征; 轨道33为p型Rydberg轨道特征.

图3 手性S-Ser分子的NTOs
2.2.2 手性S-Ser分子的自然轨道跃迁 以手性S-Ser分子S7激发态为例, 计算自然轨道跃迁(NTO)并进行可视化分析, 结果如图3所示. 由图3可见, 轨道28为N5原子的孤对电子轨道, O13原子的孤对电子对C1—C4和C1—C8的σ键略有贡献, 但可忽略不计; 轨道29为p型Rydberg轨道特征. 因此, 手性S-Ser分子S7激发态激发模式为n→p型Rydberg激发, 与图2的分子轨道(MO)结果相符, 因此可推断S7激发态的激发模式为n→p型Rydberg激发, 二者对比可判断该体系激发态的激发模式, 并可推断其他激发态的激发模式.
2.3 S-Ser手性负二价离子的单重低激发态特性
2.3.1S-Ser2-轨道跃迁的单重低激发态特性 手性S-Ser分子和离子的S7激发态电子跃迁轨道列于表4.
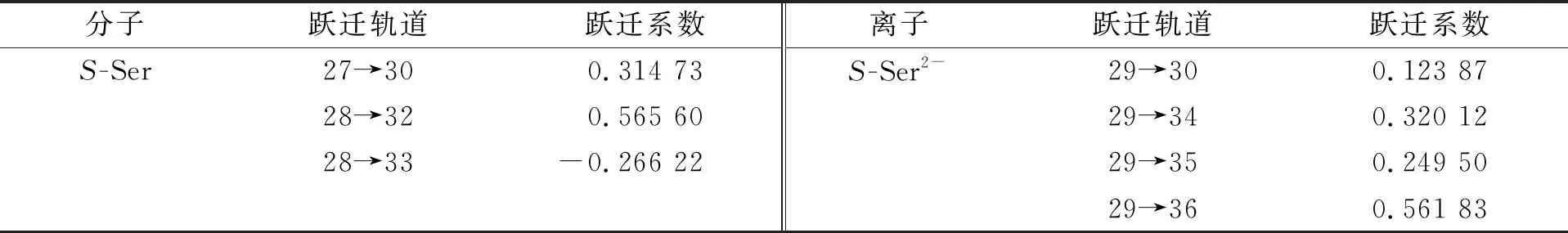
表4 手性S-Ser分子和离子的S7激发态电子跃迁轨道
以S7激发态为例,S-Ser2-的S7激发态MO如图4所示. 由图4可见: 与S-Ser分子的跃迁轨道相比, 其轨道数增加4条, 轨道系数变化较大; 轨道29为s型Rydberg轨道特征, 轨道36为羧基C—O键的反π(π*)轨道.
2.3.2S-Ser2-的NTOS-Ser2-的S7激发态NTO计算结果如图5所示. 由图5可见, 与手性S-Ser分子的跃迁轨道相比, 其轨道由28向29跃迁变为由29向30跃迁, 且跃迁系数发生明显变化; 轨道29为s型Rydberg轨道特征, 轨道30为羧基C—O键的反π(π*)轨道. 因此手性S-Ser2-的S7激发态激发模式为s→π*激发, 与图4结果基本相同, 仅个别原子的贡献略有差别, 但可忽略不计.
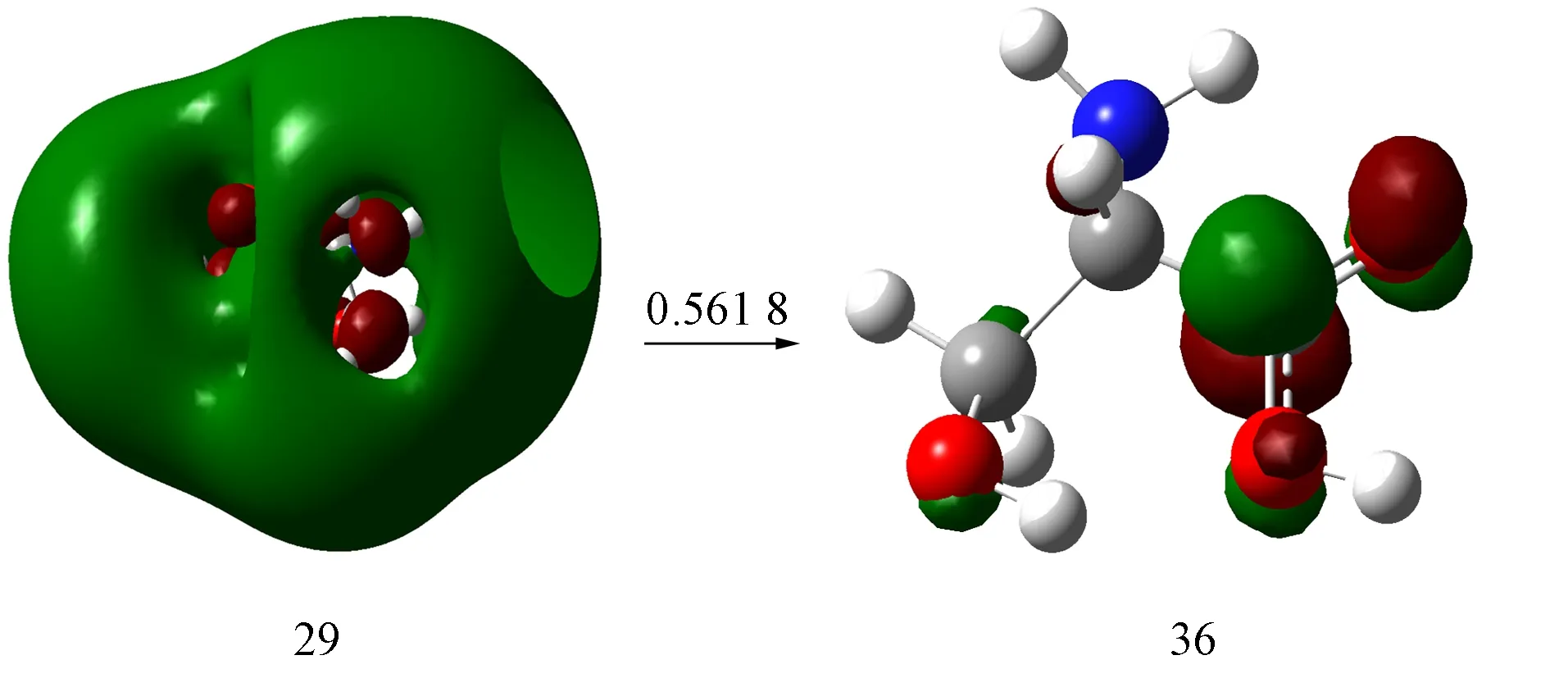
图4 S-Ser2-的S7激发态MO
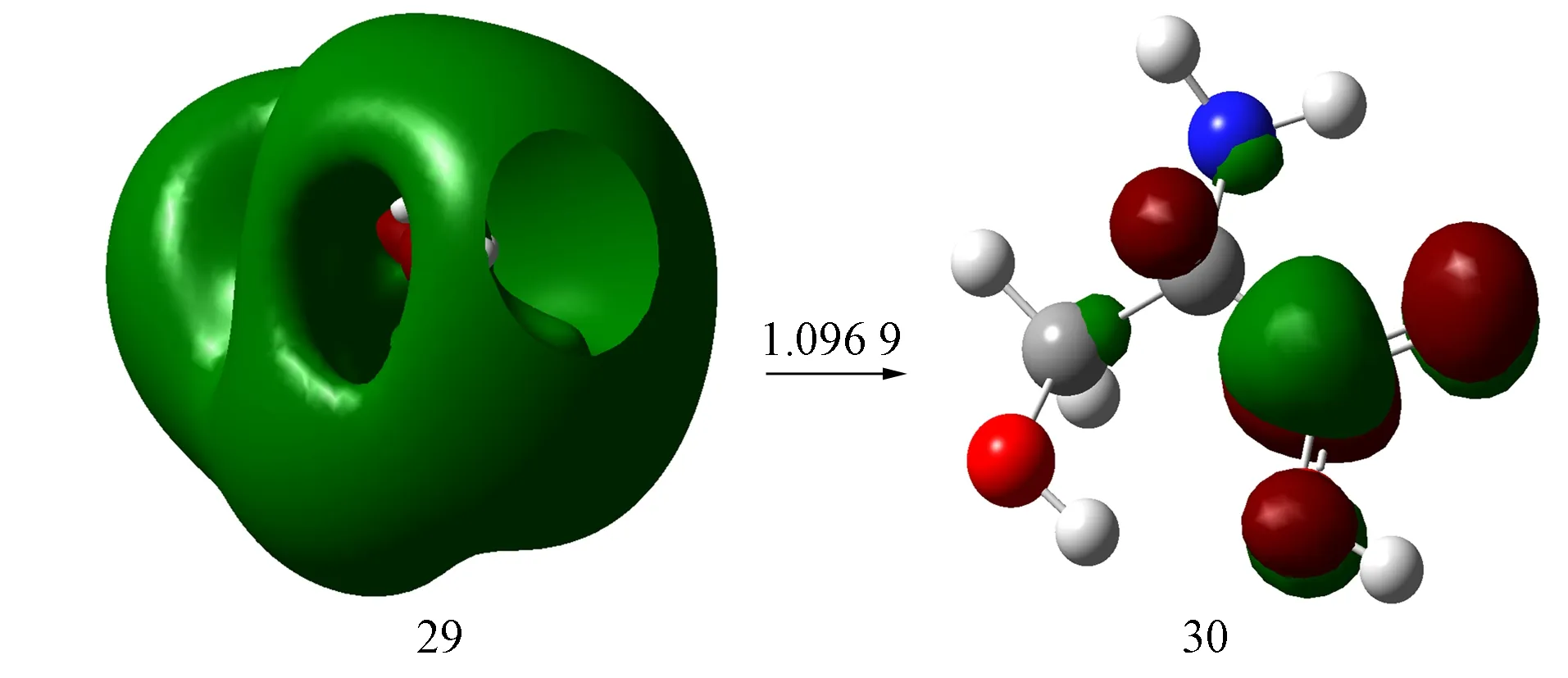
图5 S-Ser2-的S7激发态NTO
综上, 本文可得如下结论: 手性S-Ser分子和S-Ser2-结构参数随分子体系俘获电子数目的增加而发生变化, 其中手性分子键长R变化最明显, 手性分子俘获电子直接影响其原子电荷分布; 基于含弥散函数的基组, MO结合NTO特征分析法可较好地判断手性S-Ser分子和S-Ser2-激发态电子的激发特性, 对Rydberg激发特征指认效果较好.
