Ptn-石墨烯体系氧还原反应的第一性原理计算
2020-07-18曹妙聪
曹妙聪, 徐 强, 师 帅
(1. 长春工程学院 勘查与测绘工程学院, 长春 130021; 2. 吉林大学 材料科学与工程学院, 长春 130012)
由于贵金属Pt的储量有限, 因此其作为质子交换膜燃料电池中最有效的电化学催化剂受到限制[1]. 为保证催化剂的活性并减少Pt的用量, 需提高Pt的分散度并增加选择性和稳定性, 或寻找有效的Pt合金[2]和非贵金属[3-4]. 文献[5-11]研究表明, 以石墨烯表面为基底, Pt团簇吸附在其表面, 可提高催化剂的分散度, 并增强了选择性和稳定性. 对该稳定的复合体系, Lim等[8]研究了O2在Pt-石墨烯体系上的吸附能及吸附方式, 并得到了Pt13团簇-石墨烯上的氧还原反应(ORR)路径; Kaukonen等[7]研究了在Pt-石墨烯体系上ORR的中间产物, 但ORR开始前的O2如何从无穷远处吸附到石墨烯体系表面, 再从吸附的O2或O原子开始反应以及O2如何在Pt-石墨烯体系表面吸附和脱附的研究目前尚未见文献报道. 基于此, 本文在Pt-石墨烯体系上ORR的中间产物基础上, 研究Pt2-石墨烯和Pt4-石墨烯体系上ORR的中间产物和能量变化, 并计算两种竞争反应路径中H2O分子的最低脱附能垒.
1 模拟计算
采用基于密度泛函理论的缀加投影波(PAW)方法, 用广义梯度近似(GGA)-PBE表示相互作用的电子交换关联能. 平面波截止能为400 eV, 总能量收敛在每个原子1 meV. 在计算过程中, 考虑原子自旋. 首先, 构建单层六方石墨烯(4×4)超晶胞结构, 以4×4×1的单空位石墨烯为基底吸附Pt原子和团簇; 其次, 优化石墨烯结构, 其晶格常数为0.246 nm, 与实验值相符. 4×4超胞的Brillouin区用Г中心的K点网格10×10×1. 为避免石墨烯层间的干扰, 相邻两层石墨烯间的距离设为2 nm. 所有计算均在VASP软件包[12]内完成.
2 结果与讨论
2.1 O2在Pt-石墨烯体系中的吸附及氧化还原反应
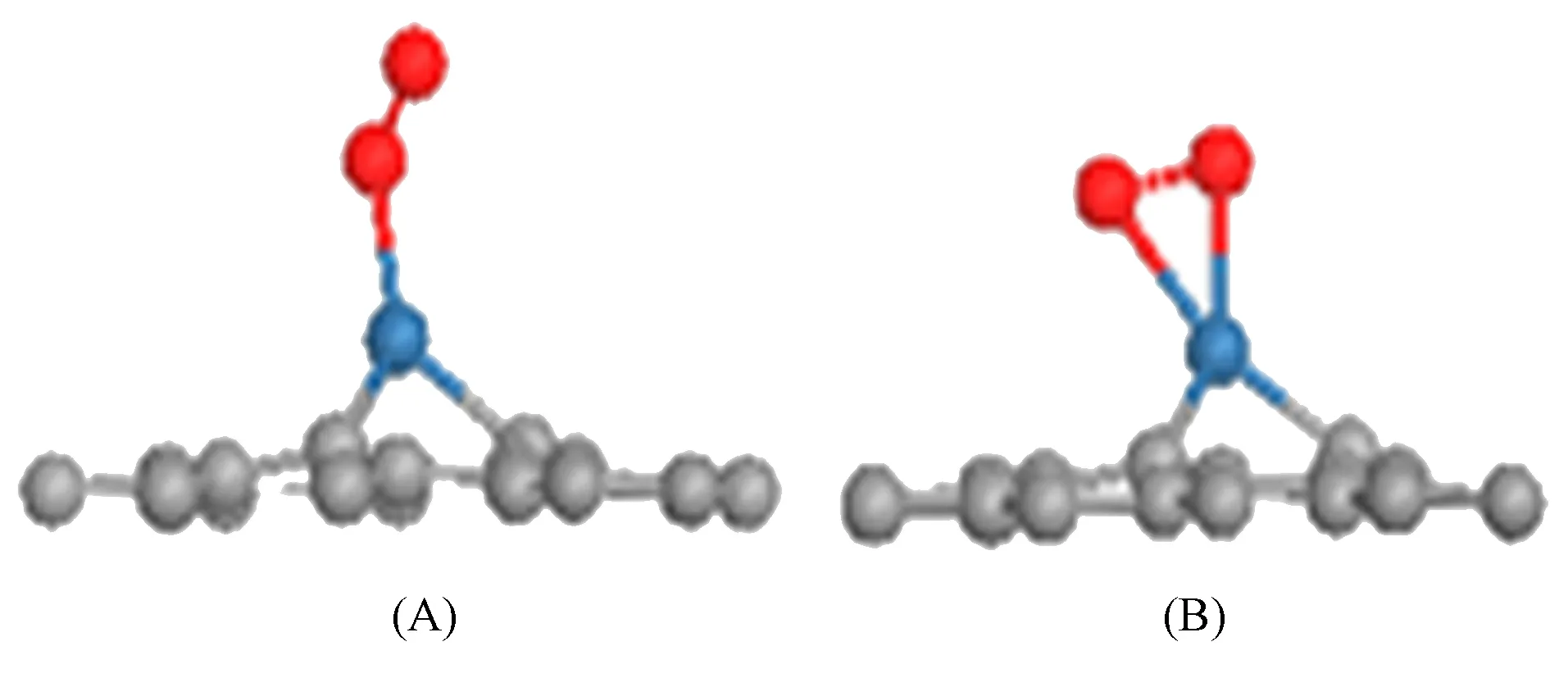
图1 O2在Pt-石墨烯体系中的不同吸附方式
O2在Pt-石墨烯体系中的不同吸附方式如图1所示, 其中: 红色球表示O原子; 蓝色球表示Pt原子; 灰色球表示C原子. 由于Pt-石墨烯体系是优化后的晶胞结构, 因此可得到稳定吸附的O2. 图1(A)为一个O原子与Pt直接相互作用, O—O键长为0.132 nm, O—Pt键长为0.194 nm, 吸附能为-1.581 eV; 图1(B)为两个O原子均与Pt直接相互作用, O—O键长为0.143 nm, O—Pt键长分别为0.197,0.214 nm, 吸附能为-1.54 eV, 与真空中O2分子相比, O—O键明显拉长, 其相互作用变弱. 两种吸附方式的O—Pt键长均小于0.2 nm, 表明O2与体系间形成了稳定的化学吸附. O2吸附到Pt13-石墨烯的吸附能为-2.3 eV, 吸附更稳定[8].
本文研究图1(B)的吸附方式. O2在Pt-石墨烯中的吸附过程及其能量如图2所示, 其中: 红色球表示O原子; 蓝色球表示Pt原子; 灰色球表示C原子. 选相对稳定的吸附位置d=0.57 nm, 此时吸附能为0.072 eV, 表明O2与Pt-石墨烯体系作用较弱. 下面分两种方式研究吸附情况, 将O2中的两个O原子同时直接吸附到Pt-石墨烯体系, 如图2(A)所示. 图2(B)为第二种吸附过程. 由图2(B)可见, 两个O原子分别靠近Pt-石墨烯体系, 第一个O原子随着d的减小, 最终形成稳定吸附的O—Pt-石墨烯体系, 为间接吸附方式. 另一个O原子逐渐靠近O—Pt-石墨烯体系, 最终形成稳定吸附的O2, 如图2(C)所示. 由图2(D)可见, 直接吸附需0.7 eV的能垒, 而间接过程需要的能量较高, 这是由于O2分子键断裂较难, 使其吸附过程需克服较高能垒6.16 eV所致. O2的吸附过程共放出1.612 eV的能量. 吸附过程的逆反应为O2的脱附过程, 间接脱附比直接脱附难, 但两个脱附过程均需克服至少为2.32 eV的能量. 因此O2—Pt-石墨烯吸附体系较稳定, ORR从吸附的O2开始较合理.
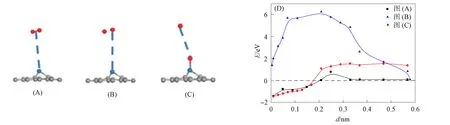
图2 O2在Pt-石墨烯中的吸附过程及其能量
在Pt-单空位石墨烯体系上, 分别吸附O2,—OOH,—OH和H2O并对吸附后的体系进行结构优化, Pt-石墨烯体系中ORR的中间态如图3所示, 其中: 红色表示O原子; 白色表示H原子; 蓝色表示Pt原子; 灰色表示C原子. 由图3(A),(B)可见, O2从无穷远处接近Pt-石墨烯体系, 逐渐形成O2—Pt-石墨烯稳定吸附体系. 由图3(C)可见, 随着H原子的吸附形成—OOH. 由图3(D)可见: 当第二个H原子吸附时, 形成两种中间产物, 其中一种形成V字形的—OH基团, O—Pt键长为0.201 nm, O—Pt—O键角为77.30°; 当吸附H原子后, 将脱附一个H2O分子, 形成O—Pt-石墨烯体系, O—Pt键长为0.179 nm, 且竞争过程存在于上述两种反应中. 由图3(E)可见, 当第三个H原子吸附时, 形成了稳定吸附的OH—Pt-石墨烯体系, 并伴随一个游离的H2O分子. 由图3(F)可见, 第四个H原子吸附到体系, 吸附于Pt原子的H2O分子为亚稳态, 易脱附. 不同中间产物的能量列于表1. 由于
Ead=E2-E1-0.5E(H2),
其中:E(H2)为氢分子的能量;E1和E2为H原子吸附前后体系的能量. 因此H原子吸附过程均为放热反应, 且吸附体系稳定, 仅在第二个H2O分子脱附时需克服能量0.426 eV, 总反应释放能量5.317 eV.
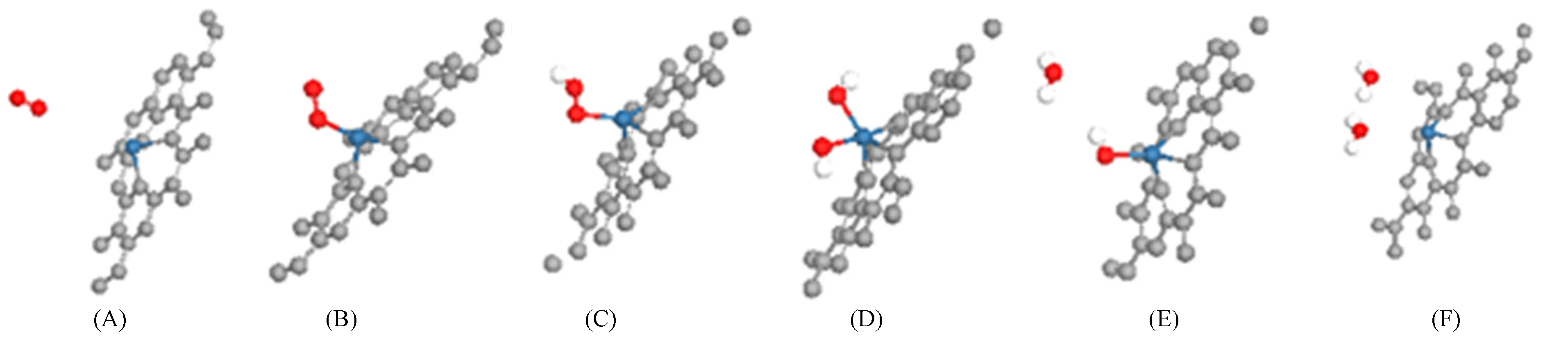
图3 Pt-石墨烯体系中ORR的中间态

表1 Pt-石墨烯体系中ORR的中间态能量
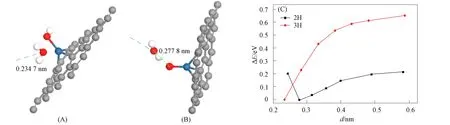
图4 Pt-石墨烯体系中H2O的脱附过程
下面考虑图3(C)~(E)的两种竞争反应, 找到H2O分子的易脱落点. Pt-石墨烯体系中H2O的脱附过程如图4所示, 其中: 蓝色虚线表示脱附路径; 黑色和红色线分别表示第二个和第三个H原子吸附时H2O分子脱落的能量变化. 选择两种稳定的吸附体系, 以OH—Pt-H2O和Pt—O-H2O为脱附起点, 图4(A),(B)为优化后的结构体系. 由图4(A)可见, 当以V字形—OH吸附时, 第三个H原子吸附时H2O分子脱附, H2O分子中的O—Pt键长为0.234 7 nm, 每选定一个新位置均固定除H2O分子外的全部原子, 但固定H2O分子中氧原子的z轴坐标. 图4(B)为H2O分子脱附示意图, 当第二个H原子吸附时, O—Pt键长为0.277 8 nm. 图4(C)为H2O分子脱落时的能量变化. 定义最优位置的吸附能为E0, 变换位置的吸附能为EN, 纵轴为ΔE=EN-E0, 横轴d为H2O分子中O和Pt原子间的距离或两个O原子间的距离. 图4(B)中H2O分子脱附所需能量仅为0.212 eV, 由于H2O分子与Pt间距离小于H2O分子吸附的最优位置与Pt间距离, 因此, 当H2O分子靠近Pt原子时, 系统不稳定. 图4(A)的脱附过程所需能量为0.6 eV, 脱附较难. 但两种反应路径所需能量相差较小, 可作为竞争反应, 同时存在于ORR过程中.
将两个—OH基团同时连接在Pt原子上, 初始结构见图3(D), 考察中间产物OH的脱附路线及脱附势垒, 结果如图5所示, 其中: 横坐标为O—Pt间距离; 纵坐标为体系与稳定吸附时体系的能量差. 结果表明, OH脱附需克服2.56 eV的能垒, 明显高于H2O分子脱附所需能量.
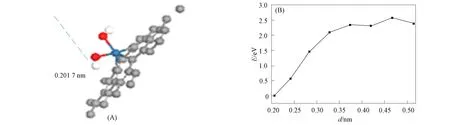
图5 OH脱附及能量变化过程
上述结果表明, 从真空中的O2分子吸附到Pt-石墨烯体系需克服能垒0.7 eV, 为放热反应, 得到了稳定的O2—Pt-石墨烯体系, O2和O原子均较难脱附. 当第一个H原子吸附时, 将获得—OOH基团, 并释放1.246 eV的能量, 当第二个H原子吸附时, 出现两个竞争反应过程, 直接脱附一个H2O分子需克服能垒0.2 eV. 若不脱附H2O分子, 将释放能量2.472 eV, Pt原子连接两个OH, 并于第三个H原子吸附时脱附H2O分子, 需克服0.6 eV的能量. 当继续吸附H原子时, 第二个H2O分子脱附需吸收0.426 eV的能量. 通过对比可见, H2O分子比O2和—OH基团更易脱附.
2.2 Pt2-石墨烯和Pt4-石墨烯体系上ORR的中间产物
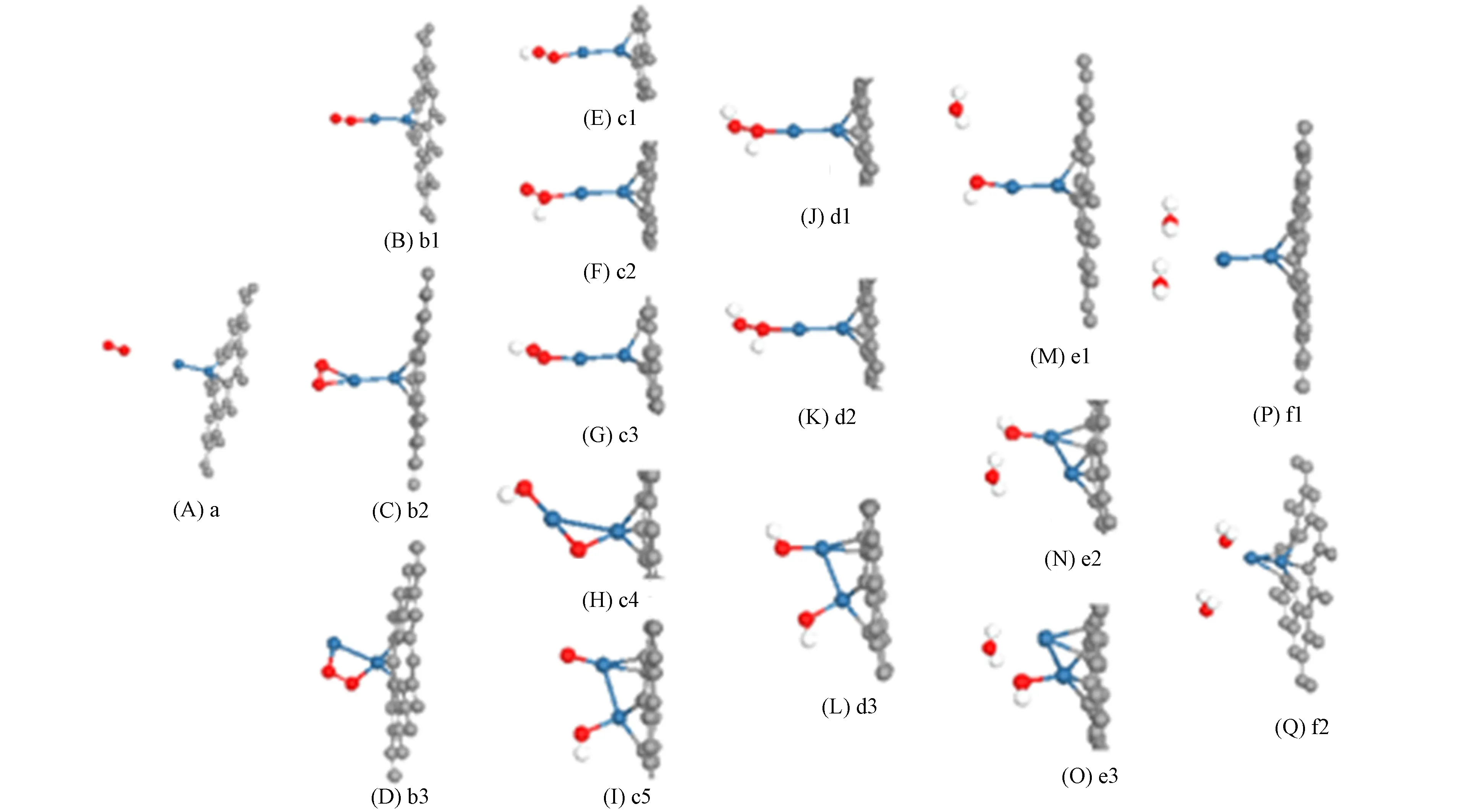
图6 Pt2-石墨烯体系中ORR的中间产物
Pt2-石墨烯体系中ORR的中间产物如图6所示. 选取O2距离Pt原子0.8 nm的位置作为ORR的初态, 如图6(A)所示, O2分别由顶点吸附的(b1)和(b2)及桥位吸附的(b3)3种吸附方式吸附于Pt2-石墨烯体系. 由图6可见: 当第一个H原子吸附时, 产生5种构型, 其中不稳定构型为图6(F); 当第二个H原子吸附时, 形成3种构型, 图6(E)构型中另一个O原子吸附H原子获得图6(J)构型, 图6(G)构型中另一个O原子吸附一个H原子得到图6(K)构型, 图6(H),(I)构型中吸附H原子后得到图6(L)构型, 吸附的能量变化列于表2, 其中括号内能量值表示由不同初始态变到相同态所需的能量.

表2 Pt2-石墨烯体系中ORR的中间态能量
由表2可见: 由e1脱附H2O分子需释放2.478 eV的能量, 由e2,e3脱附H2O分子需吸收能量; 当第二个H2O分子脱附时, 将释放能量0.512 eV, 如图6(D)所示; 当图6(Q)所示的构型脱附H2O分子时, 需吸收能量, 同时改变了体系Pt2-石墨烯的结构. 因此a→b1→c1→d1→e1→f1是催化ORR最可能的反应路径, 能量逐渐降低, 该反应释放5.144 eV能量, 与Pd(Pt)-石墨烯的H2O分子脱附能为5.1 eV相符[7].
Pt4-石墨烯体系中ORR的中间产物如图7所示. Pt4-石墨烯体系中ORR的中间态能量列于表3, 其中括号内外能量值表示由不同初始态d1和d2变到相同态e所需的能量. 由表3可见, a→b1→c1→d1→e→f或a→b2→c3→d2→e→f为ORR的可能路径, 需克服的最大能垒为0.57 eV. 与Pt2-石墨烯体系不同, 其H2O分子脱附为吸热过程, 总反应释放5.137 eV能量. 对Pt-石墨烯、 Pt2-石墨烯和Pt4-石墨烯催化ORR释放的能量分别为5.317,5.144,5.137 eV, 表明在反应过程中所释放的能量差距较小.
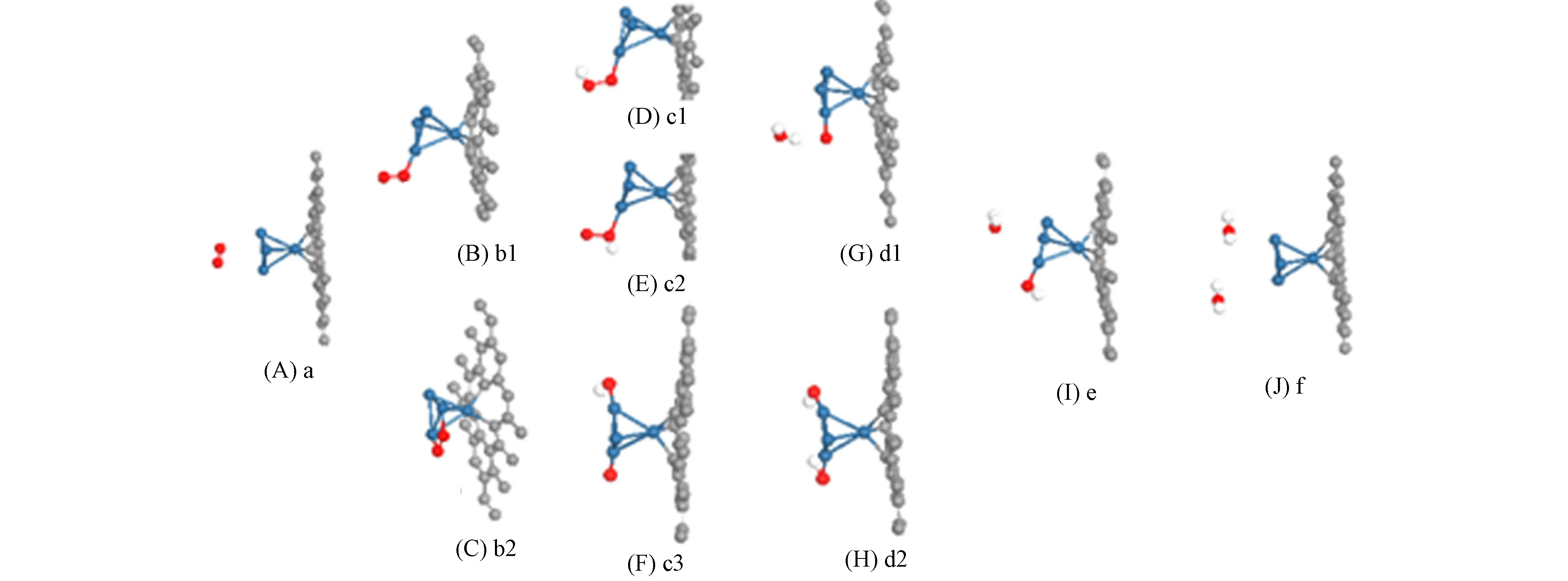
图7 Pt4-石墨烯体系中ORR的中间产物
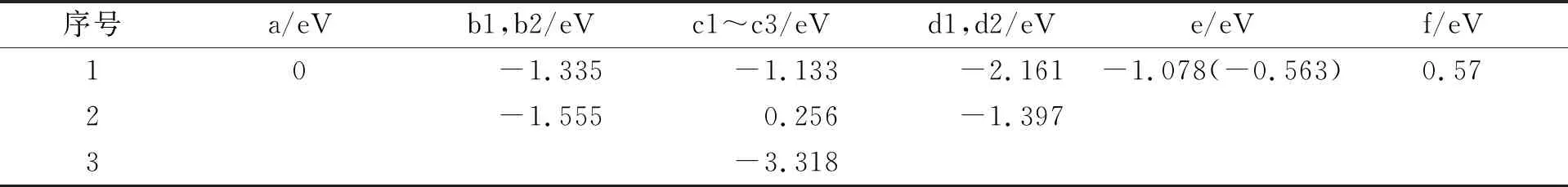
表3 Pt4-石墨烯体系中ORR的中间态能量
综上, 本文用第一性原理分析了O2分子与Pt-石墨烯体系间的作用, O2在体系上吸附容易, 脱附较难, 拆分为单个O原子单独吸附, 吸附势垒较大. 因此, 将吸附的O2作为ORR起点. 通过优化结构得到了Pt-石墨烯、 Pt2-石墨烯和Pt4-石墨烯体系中ORR的中间产物及其相应的能量变化和最可能的反应路径. 结果表明: Pt-石墨烯体系ORR的H2O分子脱附为吸热过程, 其他过程均为放热过程; Pt2-石墨烯体系的反应过程均为放热过程; Pt4-石墨烯体系的第一个H2O分子脱附可能为放热过程, 第二个H2O分子脱附为放热过程. 上述3种体系反应释放的能量均约为5 eV, 表明催化体系未因ORR而使结构发生变化, ORR也不会因催化体系的不同而吸收或释放更多能量, 体现了催化的本质.
