碳酸盐晶格硫研究进展
2020-07-02李鑫曹红耿威孙治雷张喜林闫大伟秦双双徐翠玲张现荣翟滨王利波
李鑫,曹红,耿威,孙治雷,张喜林,闫大伟,4,秦双双,4,徐翠玲,张现荣,翟滨,王利波
1.自然资源部天然气水合物重点实验室, 中国地质调查局青岛海洋地质研究所,青岛 266071
2.青岛海洋科学与技术试点国家实验室海洋矿产资源评价与探测技术功能实验室,青岛 266071
3.中国地质大学(北京),北京 100083
4.中国海洋大学,青岛 266100
碳酸盐晶格硫(carbonate associated sulfate,简称CAS),系指在碳酸盐成岩过程取代碳酸盐离子并保存在晶格中的硫酸盐[1]。它广泛存在于成岩作用形成的碳酸盐岩中,在生物成因的碳酸盐岩中也常见,包括软体动物和腕足类动物的壳体,有孔虫等[2-3]。从微晶方解石到生物成因的碳酸盐岩,CAS的含量范围通常为 0~10 000×10−6[4]。
CAS是重建古海洋环境硫循环的重要载体,前人对CAS的成因做了很多细致工作。硫酸盐作为海水中主要的阴离子,能够影响地球表面化学过程和气候变化,在生物地球化学中扮演着重要的角色[5],因此是海水硫循环和古环境研究中的关键对象,而硫酸盐的氧、硫同位素在重建海水古环境方面更是关键的评价指标[6-7]。过去常用蒸发岩作为海水古环境的评价体系,但由于它的沉积受时间限制,在地层中并不连续,且优先沉积在浅水环境下,易受后期成岩作用和流体的影响,所以很难作为理想的载体[8-9]。CAS存在于碳酸盐岩中,而碳酸盐岩在整个地质历史时期连续分布,此外CAS在进入晶格的过程中没有明显的同位素分馏,所以很好的保存了当时沉积环境的信息。碳酸盐晶格硫在20世纪60年代被发现并使用,起初对其成因和来源有很多的解释和猜测,Milliman等推测CaSO4可能是珊瑚骨架中含量很少的一部分[10],后来Mackenzie等认为CAS只存在于棘皮动物和海洋骨架碳酸盐的有机质中[11],Takano基于红外光谱实验结果提出海洋生物碳酸盐中的硫以硫酸盐取代碳酸盐的形式存在于晶格中[12]。Burdett等提出CAS不仅包括晶格中的硫,也包括流体包裹体中的硫、生物多糖蛋白中的硫、吸附相中的硫,他还认为不管什么形式的硫酸盐,其同位素组成都可以代表古海洋环境同位素组成[13]。Pingitore等利用X射线吸收光谱法发现:CAS不仅存在于海洋生物碳酸盐岩中,在成岩作用形成的碳酸盐岩中同样存在;此外CAS只存在于碳酸盐晶格中,而且CAS的同位素组成可以代表当时的沉积环境[14-16]。由于CAS具有这两种重要性质使CAS在古环境恢复方面被广泛使用:广泛存在于各种类型的碳酸盐岩的晶格中;对沉积环境信息有很好记录和保存作用。
近年来,随着同位素测试手段的不断发展,测试精度也随之提高,人们对于地质体中氧、硫同位素理解也大大加深了,使CAS的具体应用和解释能力也有了质的飞越。本文系统梳理了当前已有的研究成果,主要围绕CAS的影响因素和应用两方面进行归纳和总结,结合CAS在古环境恢复、湖泊和大气的环境评价、古冷泉碳酸盐岩的探测和识别等方面的研究趋势,尝试提出了在CAS应用过程中需要注意或急需解决的问题,希望对今后该领域的相关研究起到一定的借鉴作用。
1 CAS 的影响因素
CAS是孔隙水残留的硫酸根,主要代表孔隙水的性质,在特定的环境下CAS可以代表海水的同位素情况。受各自元素性质本身特征的影响,在同一沉积历史时期,δ34SCAS和δ18OCAS的变化可能并不同步。海水中硫的停留时间较长(约 20 Ma)[17],其硫同位素值代表当时海水处于陆表风化和火山作用输入的硫和海水硫酸盐还原输出的硫达到平衡时的水平[18]。此外,在沉积物中硫酸盐还原为硫化物过程中,直接受到微生物介导的硫酸盐还原作用(BSR)引起的碳循环的影响,所以该过程中存在较大的硫同位素动力学分馏;同时,在这一过程中微生物也优先利用包含32S的硫酸盐分子。CAS所记录的信息是海水同位素组成在沉积物中受到各种成岩作用改造后,残留的孔隙水硫酸盐同位素组成。沉积物孔隙水的硫酸盐浓度和硫酸盐还原速率会影响CAS所保存的信息。
相比硫同位素而言,CAS的氧同位素组成以一种复杂得多的形式对海洋硫循环的改变进行响应[19]。在BSR过程中,包含16O的硫酸盐分子比包含18O的硫酸盐分子更容易被还原,与32S比34S更容易还原的原理类似[20]。然而,BSR也可以导致残余硫酸盐的氧原子与环境水体中的氧原子发生交换[18,21]。这些影响都会造成硫酸盐分子富集18O,因为相对于达到同位素平衡的海水而言,海洋硫酸盐亏损18O(+9‰V-SMOW)[21]。海水和硫酸盐之间的氧原子交换受pH值和温度所控制,其同位素交换过程被CAS所记录的时间往往持续1~5 Ma[22],而这个时间尺度比硫酸盐在海水中的停留时间(约20 Ma)短得多。此外,相对于δ34SCAS,δ18OCAS对于河流输入到海洋的硫酸盐通量、硫化物歧化以及歧化物的再氧化等过程更加敏感,δ18OCAS更能反映快速剧烈的硫化物氧化途径,δ18OCAS比δ34SCAS更易受到成岩作用改造和流体的影响[23]。所以,δ34SCAS和δ18OCAS在一些地质过程中并不同步变化。然而,在有些地区,δ34SCAS和δ18OCAS又具有显著的相关性[24](图1)。这可能代表了局部的盆地环境变化,而非全球性事件。如石炭纪南美大盆地的δ34SCAS和δ18OCAS[24]。但值得注意地是,由于沉积较短,在未固结的沉积物中的δ34SCAS和δ18OCAS可能也存在较好的相关性,这种相关性主要反映沉积物现存环境的同位素水平[25]。一些学者指出,可以通过分析CAS的氧同位素组成作为依据,来判断δ34SCAS是否为原始沉积环境的硫同位素组成[26]。总之,对δ18OCAS数据进行解读时,需要通盘考虑所有可能的影响因素,结合δ34SCAS分析δ18OCAS中所蕴含的地质意义。
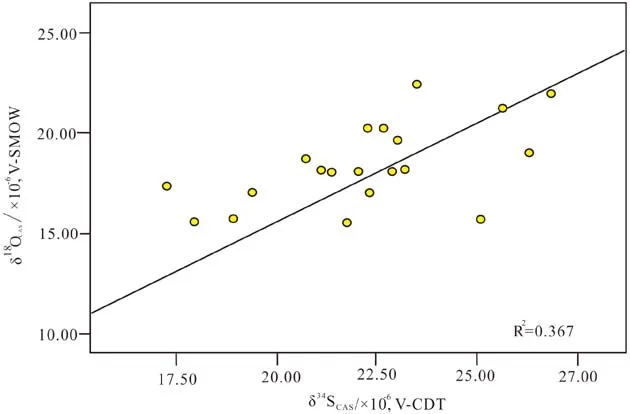
图1 δ34SCAS 和 δ18OCAS 相关性分布图(数据来自文献 [25])Fig.1 Correlation of δ34SCAS versus δ18OCAS(data from reference[25])
1.1 CAS的分析方法及对其组成的影响
总体而言,CAS的处理和分析过程较为复杂,尤其是分析测试过程可能对其同位素组成产生额外的影响,必须认真对待实验分析过程中可能存在的问题。通常,CAS提取方法包括如下3个关键步骤:①浸出水溶性的非CAS部分;②释放碳酸盐中被晶格束缚的硫酸根;③把硫酸根以硫酸钡的形式析出、固定并沉淀[2]。但对于野外样品来说,由于非CAS部分的存在,实验的难点在于如何将CAS从非CAS的部分中分离出来。常见的非CAS部分包括有机硫、酸性挥发性硫(SAS)、亚稳态的硫、重晶石、黄铁矿、石膏以及其他潜在来源的硫,它们的存在会强烈干扰对于CAS的解释,因此必须在实验中予以剔除[27]。当前主要有3种去除全岩非CAS的实验手段,分别是氯化钠溶液法、次氯酸钠溶液法和去离子水冲洗法[28]。而且对于每一种具体的实验方法,实验操作步骤和手段也有很多不同的细微之处,当前所采用的典型实验流程见表1。
由于环境恢复研究的需求驱动,CAS从提出到现在,它的预处理技术得到了迅速发展,如今已经从简单的全岩处理已经发展到微区、微量的原位分析阶段了。Burdett等首次提出CAS的分析方法,在其实验中先以5.25% NaOCl(氯氧化钠)溶液淋洗,然后用去离子水冲洗,以此去除全岩中的有机硫和不稳定的硫[12]。而后,这个方法又被进一步改进,实验时以HCl消解样品,并加入氢氧化钠调节pH(3~5),然后加入溴水促使氢氧化铁沉淀[29];与之相比,Zhang等则直接以6 mol/LHCl消解过滤,加入 5 g 的(NH4)2(OH)Cl,再加入两滴甲基橙溶液,利用HCl和NH4OH调节pH,使溶液变为红色[30];Wotte等在实验中倾向于使用NaCl溶液处理,然后用去离子水冲洗过滤,同时他们还给出了一个建议流程(图2),成为当前比较流行的全岩CAS处理的方法[2,31]。
实验处理过程中,反应发生相对迅速,因此,必须了解非CAS组分是如何影响CAS组分,才能更有针对性的去除非CAS。现有的所有全岩分析方法均首先去除可溶解的非CAS,这就意味着去除硫化物的同时,一定要避免硫化物被氧化成硫酸盐,否则非CAS组分就会很容易影响CAS本身。以最为常见的黄铁矿为例,由公式(1)可知,微生物介导黄铁矿氧化而产生Fe2+,而后,Fe2+会被氧化为Fe3+(公式2),实际上,黄铁矿也可直接发生无机氧化(公式3),而在实验室处理样品过程中公式(3)会更加普遍的发生。在这些反应里,空气中的氧气作为氧化剂参与反应,会导致生成硫酸盐的速度更快。因此,如果在提取CAS的过程中,其他组分的硫化物发氧化,最后测得的δ34S就会变成δ34SCAS和δ34Ssulfide(即硫化物中的 δ34S)的混合比值。与 δ34S类似,δ18O也会出现同样的结果,即获得的结果是δ18OCAS和提取过程中其他组分的δ18O的混合比值公式(3),而使得最后的数据难以解释。

表1 一些典型的CAS提取方法Table 1 Typical methods of extraction of CAS
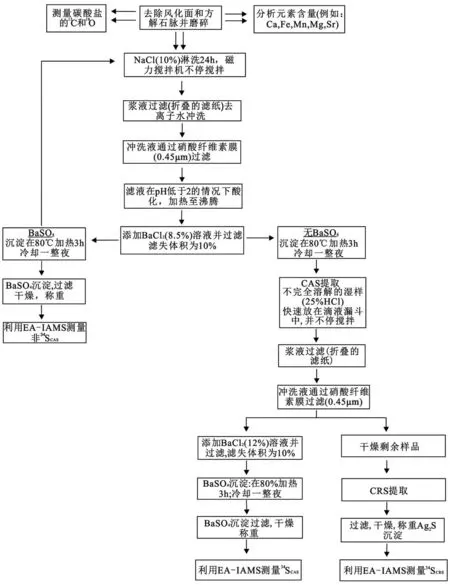
图2 CAS 提取推荐流程图(CAS 是碳酸盐晶格硫,CRS 是铬还原性硫[2,31])Fig.2 Recommended protocol for the extraction of CAS (CAS is carbonate associated sulfate,CRS is chromium-reducible sulfur[2,31])

由于实验室前处理过程中硫化物氧化为硫酸盐的广泛发生,以及对岩石样品进行研磨过程中CAS的丢失,有人提出利用质谱仪对样品进行原位分析。Paris等在其实验中使用多接收电感耦合等离子质谱仪(MC-ICP-MS)来分析CAS,这种方法可以精确分析CAS含量为nmol级别的有孔虫碳酸盐岩样品的δ34S,误差限约为0.1‰[37]。然而,该方法仍存在难以克服的不足[38]:它不能测试CAS中氧同位素组成。尽管全岩分析应用范围更广泛,从深海到滨海到潟湖沉积物都适用[39],但是实验过程必须避免非CAS部分发生氧化的现象。
鉴于此,根据不同的实验目的以及不同的岩石成分,在进行CAS提取之前,首先要进行岩石矿物鉴定和成分分析,然后再根据鉴定结果和实验目的进行CAS提取流程设计。例如,如果原岩成分含有较多的Fe2+,可以考虑加入SnCl,但要确保反应发生时是无氧环境[8];而如果样品中黄铁矿含量很高,且需要分别保留黄铁矿和CAS时,则需要在实验过程中保持全程无氧状态[8,25]。也有研究者认为CAS提取过程中黄铁矿被氧化的氧主要来自水而不是大气[40]。在CAS提取过程中,通常需要几个小时才能使CaCO3完全溶解,然后使用滤膜过滤不溶物这一过程通常在发表的文章中被省略。根据不溶物的数量和几小时的过滤步骤,致使在矿物沉淀之前,Fe3+多出几小时与黄铁矿反应,所以应该减少酸化时间,提高过滤速度(例如过滤前先用离心机把溶液与颗粒分开)。此外已有实验证明酸化过程黄铁矿会被盐酸所氧化,需要确定黄铁矿的丰度和铁的含量[40]。黄铁矿的硫同位素值较CAS的硫同位素值低得多,即使黄铁矿的δ34S远少于CAS的δ34S,但是黄铁矿氧化也会对δ34SCAS造成很大影响。当样品具有更低的δ34SCAS时,可能是黄铁矿氧化造成的影响。所以可以先用少量样品做实验确定样品中包含的活性铁含量,同时确定样品中总铁的含量和黄铁矿总量,以确定黄铁矿氧化的敏感性,然后根据实验结果来设计实验方法。但不管用哪一种方法必须对每次前处理的滤液进行测试,直到滤液中没有出现硫酸根后,再加入盐酸进行消解。
1.2 成岩作用对CAS的影响
通过分析碳酸盐岩中的CAS,可以知道碳酸盐岩沉淀时环境的氧―硫同位素组成、氧化还原变化和微生物成岩过程[41]。但迄今为止,人们对CAS在晶格中的精确位置和不同生物成矿作用中行为仍知之甚少。造成这些不确定性的原因可能是CAS在碳酸盐岩中的含量不同。如现代碳酸盐岩的CAS 浓度(1 000~10 000)×10−6,而古代的石灰岩和白云岩的 CAS 浓度仅为(0~1 000)×10−6[4]。对于后者,原因可能是古海洋本身硫酸盐的浓度很低,也可能是后期遭受成岩改变而使其浓度改变。相对而言,现代碳酸盐岩中CAS浓度的显著差异可能是生物代谢过程控制了碳酸盐结晶并且在这个过程中有其他物源的硫酸盐混入而导致的[42]。有学者认为,硫酸盐在进入碳酸盐岩晶格的过程中会发生程度很小的硫同位素分馏,但到目前仍未找到切实证据[43]。所以,CAS中的δ34SCAS值仍主要反映沉积环境海水的硫同位素组成。
事实上,我们认为地层中大尺度的CAS变化是早期成岩过程和晚期成岩过程共同作用的结果。原生沉积物中CAS变化反映海水硫酸盐或沉积环境的空间模式,早期成岩作用的发生会使CAS记录的信息受到不同程度的干扰[26],而晚期成岩过程涉及到沉积期后的硫酸盐进入CAS中,包括地下水和成岩作用改造的孔隙流体[41]等。虽然成岩过程可能掩盖了原始硫同位素在全球质量平衡框架中所占的比例,但它们确实包含了有关古环境和盆地历史的演化信息。
1.2.1 早期成岩作用对 CAS 的影响
在早期成岩作用的影响下,CAS可能不代表当时海水的氧、硫同位素组成,而是沉积物中孔隙水的氧、硫同位素情况。早期成岩作用是指沉积物沉积至浅埋藏过程中在沉积颗粒、孔隙水及沉积环境水介质之间发生的一系列物理、化学及生物学作用[44]。下面分别介绍不同早期成岩作用对CAS的影响。
对于所有未固结的沉积物来说,都会经历加压沉积,导致松散的沉积物压实、胶结,进而发生重结晶和新矿物生成等作用[45]。在这一过程中,热作用扮演了很重要的角色,它可能会导致成岩后的同位素信号产生不同程度的改变,而这对于CAS的影响尤为重要。Fichtner等以北极地区的双壳类(Arctica islandica shells)为研究对象,研究了经历早期成岩蚀变(热蚀变)前、后的CAS的变化。他们发现,当温度为175°C时,壳体内的有机质遭到持续性破坏,硫的氧化态和分布模式反映有机质发生分解和随后的硫迁移;而且在该温度的热蚀变中,新形成的方解石含量持续增加,硫的浓度变低,蚀变后析出的δ34SCAS比原生的δ34SCAS低[41]。在该实验中,除了从文石向方解石转变的过程中会发生硫酸盐的取代,高镁方解石也会出现被硫酸盐取代的特征,而且Mg/Ca比为0.13~0.21的方解石中的碳酸盐更容易被硫酸盐所取代[41,46]。所以,温度会导致原始硫的氧化进而造成硫的浓度和δ34SCAS的数值降低,而Mg/Ca的比值也能不同程度地影响蚀变后的δ34SCAS水平。
碳酸盐岩所处的空间位置不同,所经历的成岩变化也不一样,导致CAS出现较大的差异。Marenco等调查了美国西部三叠纪同一深度地层的石灰岩和距离陆地更近的白云岩[47]。其中石灰岩的δ34SCAS为 31‰~38‰(V-CDT),白云岩的 δ34SCAS为 25‰~31‰(V-CDT)。研究者认为造成这样的原因可能是白云岩化改造流体有海水的混入,其次可能由于在白云岩化过程发生了瑞利分馏[47]。当海水具有分层特征或硫酸盐浓度低时,其碳酸盐岩沉淀时的环境也成了影响CAS的关键因素。以新元古代陡山沱组浅水环境和深水环境为例,深水δ34SCAS的范围为11‰~20‰,而与之相比,浅水δ34SCAS的范围为25‰~45‰[29]。当然,也有学者也认为,浅水区更易受到后期环境改变而使CAS数据产生较大的差异,而不是原生环境的差异变化导致的[24]。此外深部沉积物中硫酸盐还原反应的发生也可能会导致这种变化。
海底碳酸盐岩成岩主要有两种途径:需氧有机质矿化和缺氧条件下硫酸盐还原菌发生甲烷厌氧氧化,这两种途径都会对CAS产生影响。Feng等研究不同成因机制形成的碳酸盐矿物(文石、方解石)发现不同途径形成的碳酸盐矿物,其CAS也不一样。文石在海底沉积物浅表层沉积,它的δ18OCAS/δ34SCAS值(0.53)与海水相近,方解石属于冷泉碳酸盐岩,形成于沉积物深部由甲烷扩散控制成岩过程,其δ18OCAS/δ34SCAS值(0.30)可能代表孔隙水的δ18OCAS/δ34SCAS同位素比[48-49]。此外在富有机质的氧化条件下,Fe3+发生氧化反应可导致CAS记录的信息是34SCAS和34Ssulfide的混合信号[50-51]。冷泉碳酸盐岩和富有机质环境下的有机质矿化形成的碳酸盐岩,它们的CAS可能反映孔隙水硫酸盐的浓度和海水硫酸盐遭到硫酸盐驱动的矿化过程影响后的同位素信号。只有在生活在海水中不与沉积物接触的生物成因碳酸盐矿物(如有孔虫、珊瑚和软体动物的壳体等)以及在还原环境下成岩作用形成碳酸盐岩中的CAS才能代表同时期海水的同位素水平。前者不会受硫酸盐还原菌和有机矿化的影响,后者则避免了Fe3+在氧化条件下把硫化物氧化为硫酸盐。
1.2.2 后期成岩作用对 CAS 的影响
后期成岩作用对CAS的影响也不容忽视,不同的成岩作用对CAS的影响也各不相同。
所有出露于地表的岩石都会经历“水—岩”反应的改造,导致岩石的组成、结构和构造发生改变,碳酸盐岩也不例外[52]。研究碳酸盐岩的“水—岩”反应对CAS的影响,可以更好地利用CAS作为恢复环境的指标。含碳酸盐沉积物以及碳酸盐岩在大气中经历的变化如下:①不/亚稳定矿物转化为稳定矿物;②以碳酸盐胶结物形式发生沉淀;③原生文石和高镁方解石在相对较快的时间尺度上转化为低镁方解石,使新的沉积相与大气降水环境下的化学成分达到相平衡[53]。Gill 等利用原位分析的手段研究了佛罗里达群岛遭受大气降水蚀变的海相碳酸盐岩质珊瑚,发现已发生成岩蚀变的方解石中的Sr、Na元素浓度比原生文石低5到10倍,其中δ18O值平均降低0.86‰(V-PDB)[43]。而遭受后期成岩蚀变的碳酸盐岩中的CAS的浓度比原始值的平均降低12倍;相比而言,原生的文石,次生的方解石和原始的海水的δ34SCAS值却显示出了很强的一致性[43]。在佛罗里达群岛样品中,CAS的δ34SCAS值为20.6‰~22.6‰,而同时期海水的值为20.8‰~22.0‰[54],表明保存在碳酸盐岩中的CAS对于后期成岩作用有很强的“缓冲”作用。所以,可以研究同期海水的硫同位素信息。但受后期成岩蚀变的改造,CAS的浓度又明显发生了降低。研究者推测,这是因为大气流体中硫酸盐的含量很低,故后期形成的低镁方解石中的CAS浓度远低于现在海洋中形成的原始文石的CAS的浓度[43]。虽然如此,遭受成岩蚀变的岩石中的CAS的δ34SCAS与海水δ34S的值仍呈现出很强的一致性。研究者认为,大气成岩流体中的硫酸盐的来源主要是原生文石中硫酸盐的溶解。因此,大气降水成岩作用对CAS的影响主要表现在两个方面:①降低CAS的浓度;②降低δ18O值,但对于δ34S值影响不大。
埋藏在深部地层碳酸盐岩会受到矿液和大气水的双重影响,而这种影响会被CAS所记录。以美国伊利诺斯盆地(Illinois Basin)寒武纪地层为例,对3个连续地层的δ34SCAS和δ18OCAS的值测定后,发现δ34SCAS的值基本一致,但是δ18OCAS的值却呈现较大变化。研究者认为可能的原因是靠近密西西比河谷型矿床的地层,受到成矿流体的改变和水岩作用的影响,虽然δ34SCAS值未发生明显变化,但δ18OCAS值却表现出了混合同位素的信号,代表了后期成岩成矿流体的改造作用[32]。因此,在深部地层,CAS可能会受到地层中成矿流体的影响,导致CAS的δ18OCAS值发生改变,但δ34SCAS的值则可能不受影响。
对于来自同一样品的不同矿相组份,δ34SCAS也各不相同[55](图3)。其中,生物碳酸盐岩可以精确的记录原始沉积环境的地质和地球化学信息,而样品中的泥晶和微晶组分则可能代表了后期成岩变化和孔隙流体的改造作用[56]。以晚奥陶到早志留安提科斯提岛为例,腕足类动物中的δ34SCAS为21.15±5‰,而泥晶灰岩粉末δ34SCAS的范围为10‰~25‰[57],其中,研究者认为腕足动物代表了原始海水的硫循环情况[57]。由此可见,对同一块地区全岩的不同矿相组分分析,就可得到该地区全部时间序列的环境演化信息。
随着工业化的快速发展,空气污染变得越来越严重,对碳酸盐岩露头也产生了相应影响。而污染可能导致对CAS的解读得到完全错误的结论,但该问题却经常被研究者所忽略。Peng等调查研究了中国西北地区、美国西南地区和墨西哥西南地区的60个碳酸盐岩露头样本,其多来自干旱、半干旱地区和以及因燃烧含硫煤发生空气污染的区域[26]。研究发现,在干旱和半干旱地区碳酸盐岩露头中,由于大气降水淋滤作用有限而且缺乏硫酸盐还原菌对SAS进行再循环,SAS可以在其外表面和内部微裂隙中聚集。在雨季碳酸盐溶解在水中,旱季形成次生碳酸盐岩[58]。在这种情况下大气水带来的SAS锁在次生碳酸盐岩中,而这种次生碳酸盐岩很难从原始碳酸盐岩中剔除,即使经过多次水洗也会保留下来。SAS污染导致CAS所携带的更正的∆17O 信号(∆17OSAS为 0.36‰;∆17OCAS为−0.05‰)。此外δ34SSAS(1.1‰~4.5‰,V-CDT)远远低于古海水的δ34S(10‰~35‰,V-CDT),所以存在SAS污染会使整体硫同位素水平降低。SAS的δ18O值波动较大(0.7‰~29‰)[59],平均值为 13‰,而硫酸盐矿物的δ18O值一般为10‰~20‰(V-SMOW),尽管δ18O间的差距小于δ34S和∆17O,但是如果发生SAS污染也会使δ18O变小。在CAS提取过程中,有50%的SAS组份被认为是CAS,导致实验结论错误,进而对地质历史时期的硫同位素循环发生误判[60]。研究者最后建议,在进行CAS数据解释时必须进行Δ17O检测并且认真评估δ18O和δ34S的真实性,以确保实验数据的准确性[26,61]。

图3 CAS浓度和安提科斯提岛各物种同位素组成投点图[57]Fig.3 CAS isotopic composition versus inverse concentration with all Anticosti Island specimens[57]
2 CAS 对于古环境的指示意义
碳酸盐岩在地层中连续分布,而且分布范围广,在滨海、深海乃至潟湖等环境都有出露[39]。尽管CAS在碳酸盐岩中含量范围波动大,但CAS的同位素组成与层间石膏的同位素组成高度相似,且CAS对后期成岩作用有很强的缓冲作用,所以最近十几年CAS广泛用于研究古海水硫酸盐浓度和同位素组成[5,49,62]、分析湖泊水位波动及局部气候演变[63]、探究地质历史时期的生物多样性[64]以及判断钟乳石的成因和物质来源[65]等。
2.1 恢复冷泉碳酸盐岩成岩环境
CAS可以作为区分古冷泉碳酸盐岩和现今形成的冷泉碳酸盐岩的证据[48]。冷泉区海底硫酸盐还原驱动的甲烷厌氧氧化反应(SD-AOM)消耗了90%甚至更多的在海底沉积物中产生的甲烷[65-66]。当然,最近的研究已证明这一过程可能会有其他微生物或电子受体的参与[67],但是,甲烷厌氧氧化菌和硫酸盐还原菌这两种细菌占据最大反应比例[62]。当该反应发生时,其浓度、流量以及氧、硫同位素组成都会改变[42],而这一切都会被地质载体所记录,尤其是氧、硫的同位素比值。Antler等提出富甲烷的环境中的硫酸盐驱动的甲烷厌氧氧化反应(SD-AOM)具有更小的比值(0.24~0.40)[49],且该比值不受环境中的其他理化参数(如温度、压力、盐度以及硫酸盐的浓度等)所影响[49]。因此,有研究者提出可以利用重晶石来测量但重晶石在古代和现代的沉积状况不同[69],故其并不具备普适性。而与之相比,冷泉碳酸盐岩则是SD-AOM的直接产物,且出现在发生过SD-AOM的地质历史时期的各个阶段[70],因此,其CAS忠实且无间断地记录了的比值(图4)。
在SD-AOM活跃的区域,34S和18O的同位素行为不同[42],随着32S被低溶解度的金属以金属硫化物的形式固定,34S随之升高,18O也会升高,但因为反应过程中产生的中间硫化物中的氧原子能和周围海水发生交换,最后可与周围海水环境达到平衡,因而造成两种同位素比值出现较低的斜率[71]。由于SD-AOM发生的环境和区域较为广泛,从沉积物和水的界面到该界面以下都有发现[72]。如果发生在沉积物与水的界面,则说明沉淀碳酸盐的孔隙水与周围的海水相似;如果发生在较深的区域,则孔隙水硫酸盐的斜率曲线的左下端降低,因此,比其他地区的海水具有更高的δ34S和δ18O值[8](图5)。由于碳酸盐岩广泛分布在从滨海带到深海带的各个区域,其成因并不完全是由SD-AOM所驱使形成的[73],因此,其CAS的比值代表了不同的含义。如果该比值很大,则表明载体碳酸盐岩是有机质与硫酸盐反应耦合形成;相反,较低的斜率则表明载体碳酸盐岩是甲烷与硫酸盐发生还原耦合形成[48]。这也是以CAS作为寻找和区分古冷泉碳酸盐岩的证据基础。

图4 墨西哥湾 δ18OCAS 和 δ34SCAS 斜率配分图(黑色十字SW代表现代海水同位素组成[48])Fig.4 δ18OCAS versus δ34SCAS values of Gulf of Mexico seep carbonates grouped by dominant mineral composition (Black cross corresponds to δ18O and δ34S composition of modern seawater (SW)sulfate[48])
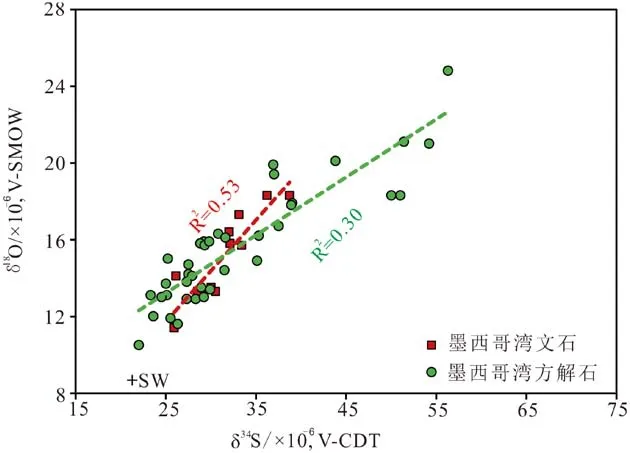
图5 现代不同地区海底 δ18OCAS 和 δ34SCAS 斜率配分图(红色为文石,绿色为方解石[48])Fig.5 δ18OCAS versus δ34SCAS values of seep carbonates from modern seafloor (Red for modern aragonite and green for ancient calcite[48])
2.2 对古海洋环境的恢复
科学家引入CAS的初衷是用来进行古海洋环境的恢复,所以该方面的研究和应用也相对成熟。Hurtgen等研究了纳米比亚Entrance和Khowarib两个剖面新元古代剖面,发现海水的硫酸盐含量非常低,而且其硫同位素记录了两次较大的正异常,可能的原因是在当时缺氧、静止的环境下,海水硫酸盐不断还原为H2S并且与Fe结合形成黄铁矿,然后被快速埋藏起来(图6a)[74]。Newton等在意大利Siusi剖面的P/Tr界线附近,利用CAS进行了高分辨的硫同位素地层学研究,发现该位置δ34S出现正偏,推测当时正处于缺氧环境,硫酸盐还原菌还原优先利用32S(图6b)[75]。He等利用高分辨率硫、氧、碳同位素的碳酸盐岩剖面,对寒武纪生物多样性进行研究:发现碳同位素和硫同位素有很好的相关性,这种相关性与寒武纪浅海环境发生的氧化作用有密切联系,而生物多样性变化是导致氧化作用变化的根本原因[64](图6c)。利用CAS对古海水环境分析,通常相对重晶石而言时空连续性好,但由于CAS实验过程复杂,影响因素较多,因此,在实际分析过程中,惯用做法是用重晶石和蒸发岩进行比较和校正[23]。

图6 CAS 高分辨率硫同位素地层学研究[64,74-75]Fig.6 The Studies of the high-resolution sulfur isotope stratigraphy of seawater sulfate[64,74-75]
2.3 CAS对钟乳石生成环境恢复
岩溶环境存在范围广泛,从陆到海都有它的踪迹,虽然地貌相似但成因却有很大的不同[76]。在岩溶环境下存在大量的碳酸盐岩,其中的CAS可以很好的揭示不同岩溶环境的成因机理。Gischler等在调查伯利兹钟乳石碳酸盐岩的成因问题时,提出了与其他研究者不同的观点,认为在这一过程中,异氧细菌尤其是硫酸盐还原菌起到了重要的作用[25],而前人一直主张这一过程主要是物理或者化学沉积造成的[77]。CAS的证据表明伯利兹生物钟乳石碳酸盐岩的 CAS 的浓度范围为(823~2 573)×10−6,在已知的海洋碳酸盐岩中属于较低范围[4]。在同沉积过程中,假设有强烈的硫还原,碳酸盐岩中硫的含量会很低,在开放环境下亏损的硫酸盐会被新流入的部分所补充,这就会造成富集重的18O和34S而不是轻的16O和32S[78]。伯利兹生物钟乳石中CAS富集相对重的18O(3.0‰)和34S(1.5‰),该模式与沉积过程中有硫酸盐还原菌参与的富集特征相同[21,79],因此,推测细菌在该地区沉积过程中起到了重要的作用。
2.4 对地质历史时期古湖泊环境的恢复
利用CAS对湖泊的研究不同于对海洋的研究,CAS对湖泊的研究相对来说很少,应用机理也较为简单。封闭的碱性湖泊有很高的碳酸盐岩含量,所以可以非常好的记录环境改变。现在可以用多种方法估计湖泊的水位和容积(如:δ18O、海岸线凝灰岩记录和生物化石),然而这些方法都存在很多局限性[80-81]。William等首次采用碳酸盐岩中的CAS浓度作为指标,研究其对于湖水化学成分和体积的响应,进而探索当时的气候变化。他们首先把CAS从自生碳酸盐岩和凝灰岩中提取出来,然后将该湖泊地质历时期中的其他数据和CAS联系起来。他发现CAS与地质历史时期湖水硫酸盐浓度变化在0~25 mM的范围有很好的线性关系,这相当于湖面发生了30 m的变化;此外数据还表明在湖水下降期间硫酸盐含量上升,这个结论与其他的研究结果相一致[82](图7,图8)。
3 展望及启示
CAS作为碳酸盐晶格中的硫酸盐,不仅具有氧、硫同位素的所有特点也兼具碳酸盐岩分布广、时空连续性好等特点[63],是很好的古环境恢复工具。而迄今,人们对于CAS在碳酸盐晶格中的确切位置以及不同形式生物矿化过程中,CAS进入晶格的途径知之甚少[41]。在迫切需要解决的与CAS有关的问题当中,未来研究焦点主要集中在提取后的同位素信号改变和环境恢复两方面,具体包括:区域CAS同位素信号与全球同位素信号响应问题;微生物矿化过程与无机矿化过程CAS的浓度和同位素信号差异问题;短期快速沉积的碳酸盐岩中CAS记录周围沉积环境信息的精确性问题;改变后的CAS同位素信号是否可以反演沉积期后作用等。这些都是当前研究者亟需解决的问题。
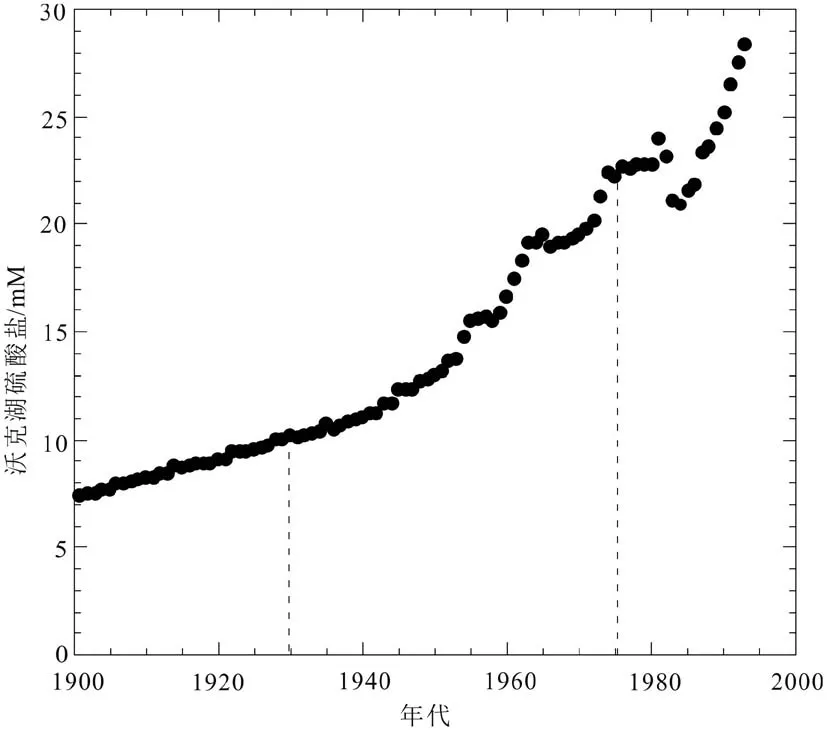
图7 沃克湖 1900―1995 年的硫酸盐浓度[82]Fig.7 Walker Lake sulfate concentration between 1900 and 1995 AD[82]

图8 沃克湖CAS和沃克湖硫酸盐浓度斜率配分图[82]Fig.8 CAS versus Walker Lake sulfate concentration[82]
就本文涉及的内容而言,至少可以提供如下两点启示:
(1)全岩分析和微区原位分析相结合,可以全面立体的反映沉积演化过程。微区分析较全岩分析而言,干扰因素多,对于反演原始沉积环境方面不如全岩分析效果好[83];但微区分析可以描绘全岩经历的连续时间尺度的CAS变化[43,64]。结合全岩分析和原位分析,可以很好的反映原始沉积环境和沉积期后发生的所有成岩蚀变。此外,也可以用两种分析方法进行相互校正,使数据更加可靠。
(2)冷泉碳酸盐岩中的CAS记录了详细的冷泉渗漏历史。冷泉区碳酸盐岩主要受甲烷厌氧氧化反应和硫酸盐还原反应控制,这两种反应不仅产生冷泉碳酸盐岩,也产生黄铁矿这类还原性硫矿物,而这两种反应受制于冷泉泄漏通量[70,84]。有学者利用陆表还原性硫储库的硫同位素值与海水硫同位素值,推测出河流输入海洋的硫酸盐通量[35]。如果今后的研究中结合这种方法,就可以大致估算古冷泉沉积期甲烷的渗漏通量。还有研究者利用CAS来进行受热液输入影响的冷泉碳酸盐岩的追踪研究[85],都是近期涌现出对CAS应用领域的创新性拓展。总之,冷泉碳酸盐岩中丰富的CAS信息,不仅可以当作区分古冷泉碳酸盐岩和现今形成的冷泉碳酸盐岩的证据[48],可能也是解开海底甲烷渗漏通量之谜的关键。