碱胁迫下榆叶梅差异基因表达分析
2020-07-01邓家林
刘 佳 田 云 邓家林
(1四川省农业科学院园艺研究所,四川 成都 610066;2农业农村部西南地区园艺作物生物学与种质创制重点实验室,四川 成都 610066)
我国川中丘陵地区为中国李(Prunus salicinaLindl.)的主要种植区,此区域的土壤广泛由石灰性紫色土组成,pH 值大多介于7.69~8.47 之间[1]。碱性土壤上生长的果树容易出现缺铁性黄化症状[2],使其生长受抑制,影响开花或坐果,甚至导致树体死亡[3]。近年来,学者们对植物如何在盐碱土地上正常生长进行了大量研究[4-5],发现选择适宜的耐盐碱植物是高效利用盐碱土壤的主要方法[5]。果树大多通过嫁接进行繁殖,所以果树的耐碱性实际取决于砧木的耐碱性。此外,砧木的品种也直接影响着果树寿命的长短、结果实的早晚以及产量的高低,因此筛选适宜的耐碱砧木是增强果树耐碱性的有效途径[6]。
榆叶梅(Prunus trilobaLindl.),又称小桃红或“鸾枝”,是蔷薇科(Rosaceae)李属(PrunusL.)落叶观花灌木或小乔木,在我国的东北、华北和西北地区广泛栽植[7-8]。榆叶梅具有很多品种,在我国已有300 多年的栽培历史[7,9]。它是一种具有2 n=8 X=64 核型的八倍体植物,具有适应性极强、耐贫瘠和低水平管理、耐寒、耐旱以及抗病性较强等特点[7,10-11]。此外,榆叶梅还是一种天然的耐盐碱性植物,在盐碱土壤(pH 值8.8,盐含量0.3%)中生长良好,与中国李嫁接后表现出亲和性好、果实早熟、产量高等特点,是川中丘陵地区李树的良好砧木[10]。
土壤盐化、碱化或盐碱化是植物常遭遇的几种非生物逆境胁迫[12-13]。研究表明,碱胁迫对植物造成的伤害远大于盐胁迫[13-14]。目前有关植物对盐碱胁迫响应的报道主要集中于盐胁迫响应机制[15],而关于植物对碱胁迫响应的报道较少,急需探明植物响应碱胁迫的各种潜在机制(如细胞生理生化与分子抗逆机制等)。近年来,多种大规模平行测序平台的飞速发展已成为探明植物响应碱胁迫分子机制的重要手段。其中以RNA-Seq 技术为基础的转录组测序,因具有耗时少、高通量、高灵敏度、操作简单、低成本等优点,在众多领域中已被大量推广运用。且该技术能够快速有效地对植物某一特定过程或阶段处理后的基因表达情况进行诊断,发掘参与胁迫响应的功能基因并预测基因的功能,使其成为目前常用于研究植物抗逆作用机制的强大手段[15-16]。RNA-Seq 技术已被应用于研究多种植物的抗碱响应机制[17-20]。
本研究采用Illumina HiSeqTM2500 高通量测序技术,对碱胁迫0、1、3、6、12 h 下的榆叶梅叶片进行de novo转录组测序,并对得到的大量转录本进行从头组装,获得大量unigenes 差异表达(DEGs),对其进行基因GO 和KEGG 富集分析,同时对7 个碱性胁迫反应相关基因进行RT-qPCR 验证,以期进一步了解榆叶梅碱胁迫分子的响应机制,同时为研究以榆叶梅为砧木的中国李在碱胁迫下的适应机制奠定分子研究基础。
1 材料与方法
1.1 试验材料及碱胁迫处理
2014年11月将榆叶梅种子(来源于新疆伊犁植物园)于沙中层积保存,2015年2月中旬,将经沙藏萌动的榆叶梅种子播种于底部直径25 cm 的带孔瓦盆。每个瓦盆中装入等量的培养土,培养土由泥炭土与草炭土按同等比例混合而成。待榆叶梅幼苗的高度长至约15 cm 后,选择大小、长势基本一致的150 株盆栽幼苗作为试验材料,对其进行碱胁迫处理。
碱胁迫处理的植株用碱液浇灌,碱液由9∶1摩尔比的NaHCO3和Na2CO3溶液混合构成,其浓度和pH值分别为50 mmol·L-1和9.11±0.104 ,碱液处理之前土壤pH 值为7.4,处理后土壤pH 值为9.0。处理组材料于晴天下午16:00 用该碱液浇灌土壤,直至钵底流出碱液开始计时,对照同期使用清水浇灌(浇灌后土壤pH 值为7.1),用透明塑料薄膜对这些材料进行避雨遮挡[21]。
1.2 RNA 提取、cDNA 文库构建及Illumina 测序
在碱胁迫处理后的0、1、3、6、12 h 分别对榆叶梅中、上部功能叶片(第2~第3 节)进行采样。每次采集的样品存放于液氮中,以备后续RNA 的提取。其中每个时间点进行2 个生物学重复,每个重复由10 株材料的叶片混合而成。本试验共10 个样本材料。总RNA 提取采用北京艾德莱(aidlab)生物科技有限公司的通用植物RNA 提取试剂盒。分别使用Nanodrop 2000 UV-Vis 分光光度计(Thermo Fisher Scientific 公司,美国)、Qubit 2.0 荧光计(Life Technologies,美国)和Agilent 2100 生物分析仪(安捷伦科技有限公司,美国)检测RNA 样品的浓度、纯度和完整性。对符合试验目的的RNA 样品用于文库制备。
提取的10 个供试榆叶梅RNA 样本送至苏州金唯智(GENEWIZ)公司进行cDNA 文库的构建和转录组测序[在Illumina HiSeq 2500 平台上采用双末端(PE)测序法进行高通量测序]。
1.3 De novo 转录组组装
对获得的FASTq 格式的大量原始测序数据(raw data)采用数据质量统计软件Trimmomatic(v0.30)[22]进行处理,去除其中的接头序列以及低质量reads(3′或5′末端质量值低于20 或者含N 的碱基;短于75 bp的序列),以此获得用于后续信息分析的clean data。使用fastqc(http://www.bioinformatics.babraham.ac.uk/projects/fastqc/)软件对供试的10 个cDNA 文库的clean data 进行质量评估,评估内容:Q20、Q30、GC 和N含量。随后,采用Trinity 软件(版本r2013-02-25)[23]实现clean data 的重新组装,再使用序列聚类软件TGICL[24]做进一步序列拼接和冗余处理,以产生更长、更完整的非冗余序列,称为unigenes。最后,使用BLAST(http://blast.ncbi.nlm.nih.gov/Blast.cgi)(e 值<1e-5)将unigenes 比对到NCBI 非冗余蛋白数据库Nr[National Center for Biotechnology Information(NCBI)non-redundant protein sequences]中,以确定unigene 的序列方向并得到最佳的功能注释。
1.4 基因表达定量与差异表达分析
每个样品获得的unigenes 均采用RSEM 软件(Ⅴ1.2.4)[25]中 的 FPKM(fragments per kilobase per million reads)方法对其基因表达量水平进行估算。采用基于负二项分布模型的Bioconductor 软件包中的DESeq(Ⅴ1.14.0)[26],对不同碱胁迫处理下样品在基因表达水平分析中得到的expected_count 数据进行处理,以此获得在碱胁迫下榆叶梅的DEGs。P值用于基因差异表达检验。使用FDR(False discovery rate)方法确定P值的阈值。按照差异显著性标准[FDR 阈值≤0.05,且|log2(fold change)|>1]筛选显著性差异表达的基因。随后,采用基于Walleius non-central hyper-geometric distribution 的R package GOseq 对差异表达显著的基因进行GO 功能显著性富集分析,P值阈值设置为0.01[27]。此外,对差异表达显著的基因采用KOBAS 软件[28]进行KEGG Pathway 富集分析,以此确定显著性DEGs 参与的最主要信号转导途径和生化代谢途径。
1.5 RT-qPCR 验证
为了验证RNA-Seq 检测到的DEGs,对其中7 个与碱胁迫相关的差异表达显著基因进行了RT-qPCR分析。用1.2 中的RNA 提取方法对碱胁迫后0、3、6、12 h 的榆叶梅叶片进行RNA 提取。使用北京Aidlab公司反转录试剂盒(TUREscript 1st Stand cDNA SYNTHESIS Kit)进行cDNA 的合成,采用40 μL 反应体系,包括500 ng 总RNA、8 μL 5×RT Reaction Mix、2 μL Oligo(dT)引物、2 μL TUREscript H- RTase/RI Mix,RNase Free dH2O 补足至40 μL。反转录反应程序:42℃、60 min,65℃、10 min,待反应结束后,得到cDNA,于-20℃保存。特异性引物情况详见表1。采用qTOWER2.2 实时PCR 系统(DBI® Bioscience,德国)进行RT-qPCR,其中所有反应均具有3 个生物学重复和技术重复。RT-qPCR 反应体系最终体积为10 μL:5 μL 1×2×SYBR ® Green Supermix(DBI ®Bioscience,德国)、0.5 μL 200 nmol·L-1正向引物、0.5 μL 200 nmol·L-1反向引物、1 μL cDNA 模板、3 μL ddH2O。RT-qPCR 反应程序:95℃预变性3 min;95℃变性10 s;60℃扩增30 s,循环39 次;溶解曲线分析,即随后将温度从60℃逐渐升至95℃,每次加1℃,每加1℃停留4 s。以ACTIN 基因作为内参基因,并通过2-△△Ct法进行数据分析。

表1 RT-qPCR 所用基因引物信息Table 1 Primer information for RT-qPCR
2 结果与分析
2.1 测序结果、组装与注释
采用Illumina HiSeq 2500 高通量测序技术对榆叶梅碱胁迫处理不同时间下的叶片进行转录组测序,共获得了595 894 216 条原始序列数据(raw reads)。将这些raw data 提交到NCBI 的SRA 数据库,其登记号为SRP093875。经过严格的质量评估和数据过滤后,共得到529 847 752 条高质量clean reads(约62.10 Gb)。每个样本clean reads 的Q20、Q30 和GC 含量分别都大于99.16%、97.69%和45.16%(表2)。结果表明获得的clean reads 具有足够高的质量用于后续研究。使用Trinity 和TGICL 软件对clean reads 进行从头组装,共获得了124 786 个unigenes,其平均长度为668.65 bp,N50 长度为1 111 bp。在组装的unigenes中,长度为200~500 bp 的unigenes 最多,有80 060个,占unigenes 总数的64.16%;其次为500~1 000 bp的unigenes,为22 426 个,占总数的17.97%(图1)。
本研究将获得的全部unigenes 与Nr 数据库进行了比对注释。结果显示,有52 691条 unigenes(42.23%)被成功注释功能。其中对注释到的近源物种进行统计,发现占总体(52 691 条unigenes)45.41%、35.30%、2.56%、1.94%和1.61%的unigenes分别匹配到梅(Prunus mume)、碧桃(Prunus persica)、苹果(Malus domestica)、川桑(Morus notabilis)和鸭梨(Pyrus×bretschneideri)中,表明榆叶梅与这几类植物具有较近的亲缘关系(表3)。此外,这些匹配到的物种除川桑外,均属于蔷薇科,也表明本研究获得的序列被正确地组装和注释。

表2 序列分析统计结果Table 2 Summary of sequences analysis
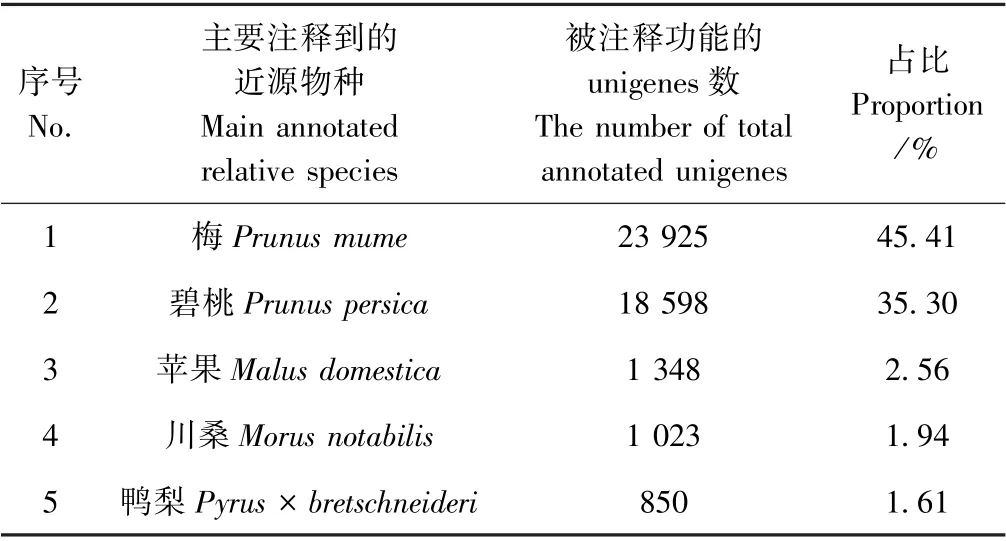
表3 被注释最多unigenes 的5 个近源物种Table 3 The five relative species annotated with the most unigenes
2.2 基因表达定量分析与DEGs 的识别
采用FPKM 法估计了每个样品的基因表达水平。在这10 个样本中,FPKM 值位于0~1 之间的unigenes占总体的20.27%~32.49%,而FPKM 值超过60 的unigenes 仅占总体的1.24%~1.57%(表4)。
碱胁迫下的榆叶梅叶片中有8 948 条unigenes(占总体的7.17%)被鉴定为显著性DEGs,其中0、186、3 211 和8 347 个显著性DEGs 分别于1 h vs.0 h、3 h vs.0 h、6 h vs.0 h 和12 h vs.0 h 比较中识别(图2),12 h vs.0 h 比较中筛选出最多的显著性DEGs。在8 948个显著性DEGs 中,3 h vs.0 h、6 h vs.0 h、12 h vs.0 h中分别有181、1 999、4 895 个DEGs 上调;3 h vs.0 h、6 h vs.0 h、12 h vs.0 h 中分别有5、1 212 和3 452 个DEGs 下调(图2)。
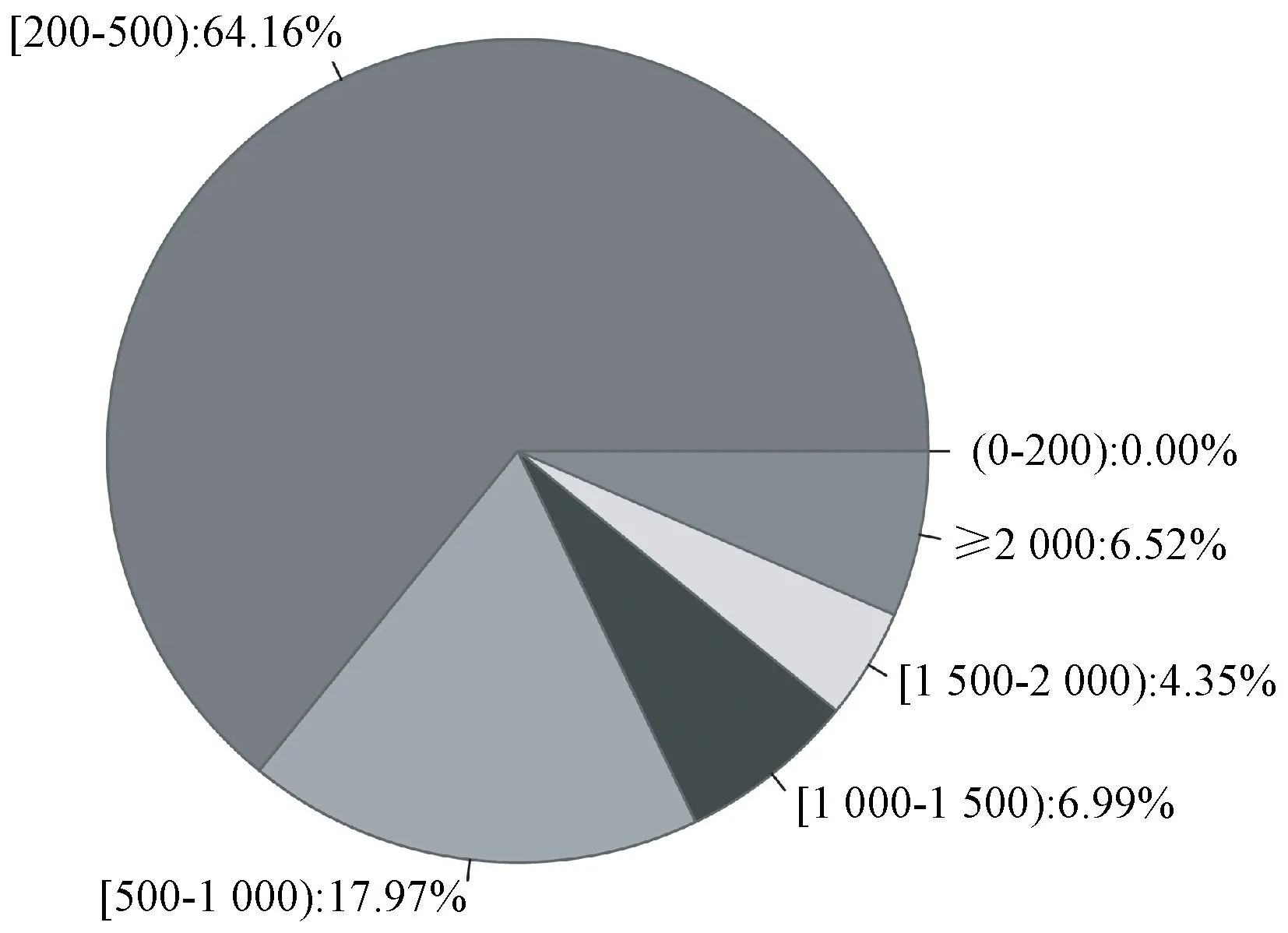
图1 组装的unigenes 的长度分布图Fig.1 Pie chart for length distribution of the assembled unigenes
2.3 DEGs 功能分类
2.3.1 GO 富集分析 为了进一步描述连续碱胁迫时间下的基因表达变化,对3 h vs.0 h、6 h vs.0 h 和12 h vs.0 h 中得到的显著性DEGs 进行了GO 功能显著性富集分析(P值≤0.01),以期找到与整个基因组背景相比,在DEGs 中显著富集的GO 条目。分析显示,GO富集包含3 个ontology,分别为细胞组分、分子功能以及生物过程。在3 h vs.0 h、6 h vs.0 h 和12 h vs.0 h中共分别识别了186、3 211 和8 347 个显著性DEGs,其中分别有0、24 和72 个显著性DEGs 被注释到GO细胞组分中;分别有9、146 和307 个DEGs 被注释到GO 分子功能中;分别有11、86 和184 个显著性DEGs在GO 生物过程中注释功能。在细胞组分类型中(图3),3 h vs.0 h 中没有显著性DEGs 注释功能,而在6 h vs.0 h 和12 h vs.0 h 中,细胞[16(66.67%);53(73.61%)]、细胞成分[16(66.67%);53(73.61%)]、细胞内部分[13(54.17%);47(65.28%)]和细胞内[13(54.17%);47(65.28%)]条目中所注释到的DEGs 数量较多。在细胞分子功能类型(图4)中,核酸结合转录因子活性[4,(44.44%)]和序列特异性DNA结合转录因子活性[4,(44.44%)]是3 h vs.0 h 中注释最多DEGs 的两个GO 条目;对于6 h vs.0 h 和12 h vs.0 h,注释DEGs 较多的条目有结合活性[70(47.95%);152(65.28%)]、催化活性[51(34.93%);106(34.53%)]、杂环化合物结合[38(26.03%)、79(27.73%)]以及有机环状化合物结合[38(26.03%);79(27.73%)]。在生物学过程功能类型(图5)中,3 h vs.0 h 中注释最多DEGs 的GO 条目为单生物代谢过程[5,(45.45%)]、氧脂代谢过程[2,(18.18%)]以及氧脂生物合成过程(oxylipin biosynthetic process)[2,(18.18%)];而在6 h vs.0 h 和12 h vs.0 h 中,代谢过程[49(56.98%);101(54.89%)]、细胞进程[28(32.56%);78(42.39%)]、有机物质代谢过程[29(33.72%);70(38.04%)]和初级代谢过程[27(31.40%);63(34.24%)]所富集的DEGs 最多。可见,随着碱胁迫处理时间的延长,更多的DEGs 被注释到功能,其中细胞组分类型中的“细胞”和“细胞成分”,分子功能类型中的“结合活性”和“催化活性”,以及生物学过程中的“代谢过程”和“细胞进程”注释到的DEGs 最多,表明榆叶梅的内部组织和器官在碱胁迫下经历了多种合成代谢过程,以此来抵御和适应碱胁迫环境。
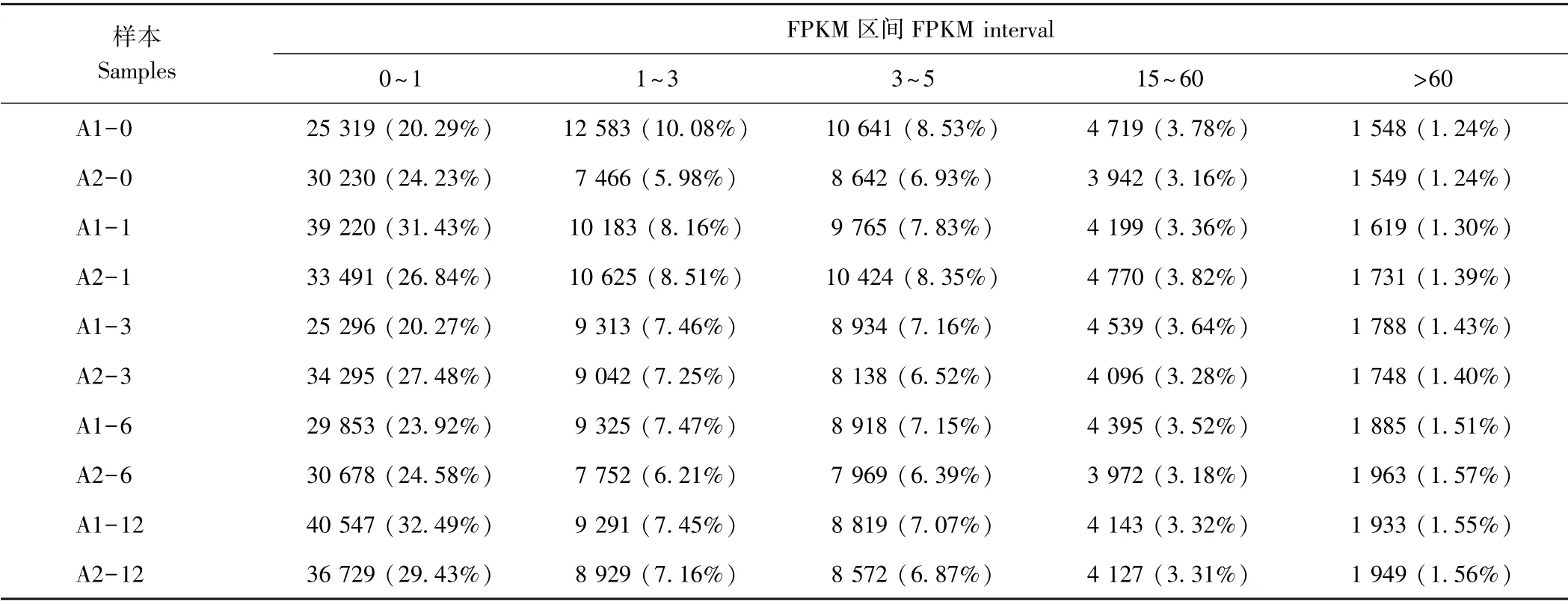
表4 不同表达水平区间的基因数量统计表Table 4 Gene number of each sample in different interval of expression level
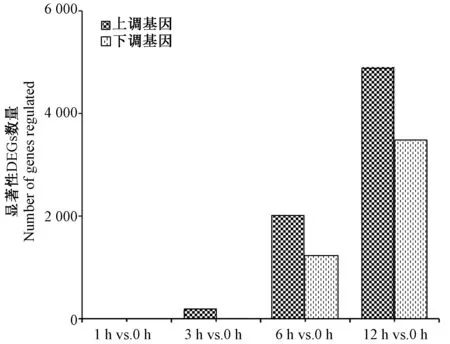
图2 碱胁迫处理下不同时间点的DEGs 数量与0 h 的对比图Fig.2 Number of DEGs at different time points after under alkaline stress compared with 0 hour
此外,在6 h vs.0 h 中,28 个基因(c57635_g1_i1、c64583_g1_i1、c64900_g1_i1、c71351_g1_i1、c72539_g1_i1、c73498_g1_i4、c73839_g1_i2、c73869_g1_i2、c74641_g1_i1、c75112_g1_i1、c76093_g2_i1、c76646_g1_i1、c78345_g1_i1、c78766_g1_i1、c79591_g1_i1、c79591_g1_i2、c79591_g1_i3、c81104_g2_i1、c82384_g1_i1、c82500_g1_i1、c83082_g2_i1、c83291_g1_i3、c83618_g1_i4、c83799_g2_i1、c86513_g2_i1、c87340_g1_i1、c87510_g1_i2 和c87918_g1_i2)比对到对刺激的应答(response to stimulus)GO 条目中,表明这些基因可能在碱胁迫响应机制中起着重要的作用。
2.3.2 KEGG pathway 富集分析 由图6可知,3 h vs.0 h、6 h vs.0 h 和12 h vs.0 h 的基因KEGG 分析结果存在着较大的差异。3 h vs.0 h、6 h vs.0 h 和12 h vs.0 h 中分别共有22、403 和1 006 个DEGs 被注释到42、41 和47 个代谢途径中。对于3 h vs.0 h,主要涉及的代谢途径分支有碳代谢途径[3,(13.64%)]、原核生物碳固定途径[3,(13.64%)]和萜类骨架生物合成途径[3,(13.64%)]。对于6 h vs.0 h,前三大富集途径依次为碳代谢途径[37,(9.18%)]、淀粉和蔗糖代谢途径[23,(5.71%)]和植物-病原体相互作用途径[21,(5.21%)]。在12 h vs.0 h 分配的47 个代谢途径分支中,碳代谢途径[77,(7.65%)]也是最富集的途径,其次是氨基酸的生物合成途径[65,(6.46%)]、淀粉和蔗糖代谢途径[53,(5.27%)]及植物激素信号转导途径[46,(4.57%)]。
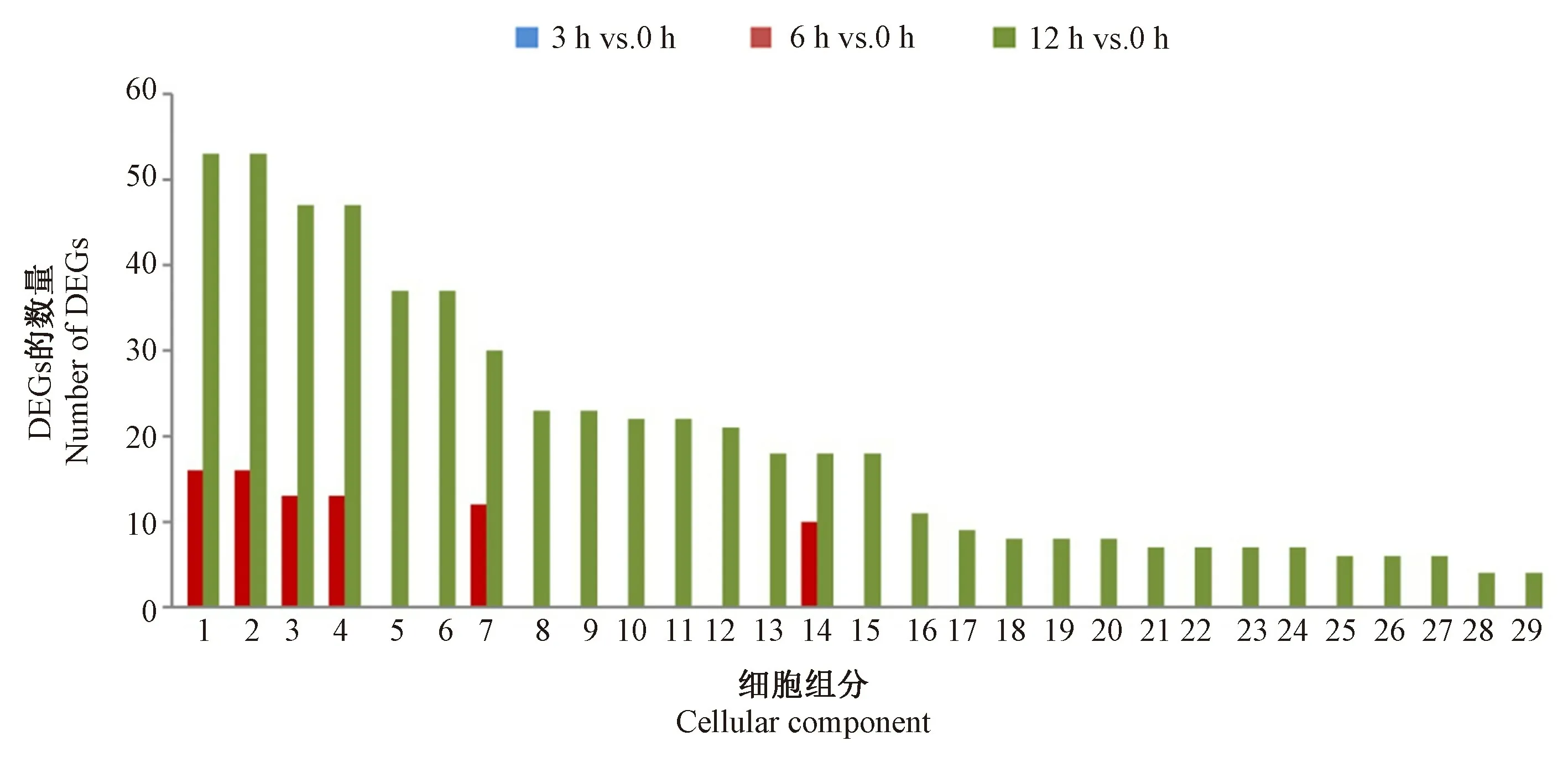
图3 3 h vs.0 h、6 h vs.0 h 和12 h vs.0 h 的GO 细胞组分功能类型对比图Fig.3 Comparative distribution of GO enrichment of terms related to cellular components for 3 h vs.0 h, 6 h vs.0 h and 12 h vs.0 h
3 h vs.0 h、6 h vs.0 h 和12 h vs.0 h 的基因KEGG 分析结果表明,榆叶梅在这些时间下均持续涉及了以下几个重要的代谢途径:碳代谢途径、光合生物碳固定途径、苯丙素生物合成、丝氨酸和苏氨酸代谢、甲烷代谢途径、丙酮酸代谢途径、乙醛酸和二羧酸代谢途径、萜类骨架生物合成途径、α-亚麻酸代谢、色氨酸代谢途径、柠檬烯和蒎烯降解途径。在碱胁迫3、6、12 h 条件下,均涉及“碳代谢途径”的相关基因为c86254_g5_i1、c86254_g2_i1、c58505_g1_i1。对这些碳代谢相关基因进行KEGG 注释表明,c86254_g5_i1 和c86254_ g2 _ i1 均为磷酸烯醇式丙酮酸羧化酶(phosphoenolpyruvate carboxylase,PEPC,EC 4.1.1.31)基因,而c58505_g1_i1 为乙酰CoA 酰基转移酶(acetyl-CoA C-acetyltransferase,AACT)基因。
对28 个比对到对刺激应答GO 条目中的基因进行KEGG 注释表明,c71351_g1_i1 是一个RNA 聚合酶相关蛋白,而c73869_g1_i2 是一个HSP20 家族蛋白。对这些基因进行COG 功能注释,发现有5 个基因比对到“翻译后修饰,蛋白质折叠和分子伴侣类基因”(c73869_g1_i2,c74641_g1_i1,c78766_g1_i1),“复制、重组和修复类基因(replication,recombination and repair)”(c82384_g1_i1),和“核苷酸转运和代谢类基因(nucleotide transport and metabolism)”(c83082_g2_i1)中。
2.4 RT-qPCR 验证
采用基因特异性引物的RT-qPCR 分析对RNASeq 检测到的与碱胁迫相关的DEGs 进行了验证。RTqPCR 验证选取了7 个候选基因(c78498_g1_i1、c80260_g1_i1、c81662_g1_i1、c84090_g1_i1、c85502_g2_i1、c85846_g1_i3 和c88652_g1_i2),并将ACTIN 基因作为数据归一化的内参基因。RNA-Seq 数据分析显示,这7 个基因均属于上调基因,且基因的大多数表达模式与通过RT-qPCR 验证得到的表达模式基本一致,且RT-qPCR 验证中这些基因在3 种碱胁迫时间处理下均呈极显著性差异表达(P<0.01)(图7)。
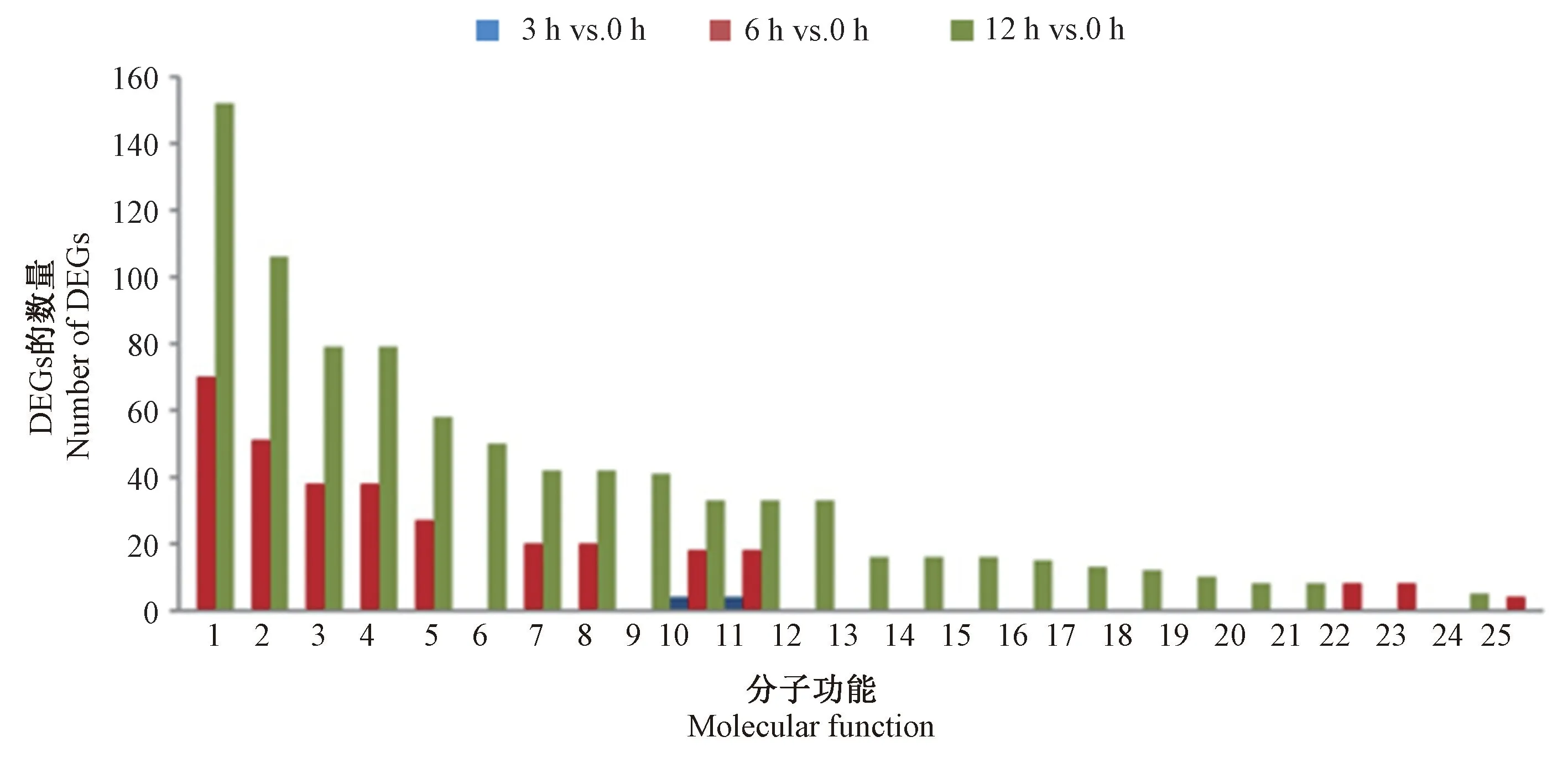
图4 3 h vs.0 h、6 h vs.0 h 和12 h vs.0 h 的GO 分子功能类型对比图Fig.4 Comparative distribution of GO enrichment of terms related to molecular function for 3 h vs.0 h, 6 h vs.0 h and 12 h vs.0 h
3 讨论
本研究运用Illumina HiSeqTM2500 技术对无参考基因组的榆叶梅碱胁迫处理不同时间下的叶片进行了转录组测序,共获得了将近5 300 万条高质量clean reads,并经过de novo组装后获得了124 786 条榆叶梅unigenes,获得的大量榆叶梅转录本序列将为榆叶梅物种提供重要的基因信息,可用于碱胁迫下重要功能基因的挖掘。本研究对碱胁迫下榆叶梅的DEGs 进行研究,发现榆叶梅叶片在12 h vs.0 h 比较中筛选出最多的显著性DEGs,这与星星草(Puccinellia tenuiflor)[29]和野生大豆(Glycine soja)[30]根中所获得的结果不一致,其星星草(Puccinellia tenuiflor)和野生大豆(Glycine soja)在6 h 筛选出的DEGs 量比在12 h 和24 h 更高,这可能是因为叶细胞响应外界刺激的速度慢于根细胞。本研究在碱胁迫3 h 下未检测到显著性DEGs,这与野生大豆(G.soja)[17]根中所得结果类似。此外,本研究3 h vs.0 h、6 h vs.0 h、12 h vs.0 h 中呈上调的DEGs 数量比下调的多,这与在多种非生物胁迫下的拟南芥转录组结果基本一致[30]。
DEGs 的功能分类将阐明其在细胞内的分子功能和生物代谢途径,对挖掘榆叶梅抗碱基因具有重要的意义。本研究中,GO 功能显著性富集分析表明,3 h vs.0 h 中只有少量显著性DEGs 注释到GO 条目中,这些GO 条目仅包括分子功能类型中的“核酸结合转录因子活性”和“序列特异性DNA 结合转录因子活性”,以及生物学过程类型中的“单-生物代谢过程”、“氧脂代谢过程”和“氧脂生物合成过程”。这些GO 条目反映了榆叶梅对碱胁迫的早期响应,以及这些条目可能诱导产生后期的级联反应。因此,这些基因在早期抗碱机制中可能起着重要的作用。本试验中,KEGG pathway 富集分析结果表明,榆叶梅叶片中代谢途径随碱胁迫处理时间的延长而有所变化。但在碱胁迫处理3、6 和12 h 条件下,榆叶梅均持续涉及了几个重要的代谢途径。其中“碳代谢途径”涉及最多的DEGs,表明榆叶梅为了应对碱胁迫需要大量的能量供应,以及碱胁迫可能主要通过改变碳代谢相关基因的表达来影响基因转录情况。本试验发现PEPC和AACT两个基因在逆境胁迫中具有重要作用。PEPC 主要在植物细胞中参与植物的光合碳同化等重要代谢途径,以及调节细胞pH 值,保持离子平衡,调节气孔保卫细胞的渗透压及运动等[31]。AACT 为萜类化合物生物合成甲羟戊酸(mevalonic acid,MVA)途径的起始酶,催化使2个分子的乙酰CoA 缩合为乙酰乙酰CoA,主要参与植物的呼吸作用等[32]。此外,一些代谢途径仅在某一处理时期涉及有DEGs。在碱胁迫处理3 h 时,发现一个特异基因(c58505_g1_i1)涉及了“钙信号途径(calcium signaling pathway)”,另一个特异基因(c89089_g2_i1)涉及了“ABC 转运蛋白途径(ABC transporters pathway)”。钙信号途径是生物体内生理生化反应的重要控制途径,研究证明其能间接影响植物的生理活动,如气孔关闭、花粉管发育,根系伸长等[33]。钙信号途径在碱胁迫处理3 h 时的特异响应,有可能与帮助榆叶梅应对碱胁迫,形成胞内钙离子浓度振荡变化,进而引发气孔关闭,抵御离子毒害有关。ABC(ATP-Binding Cassette)转运蛋白是目前已知最大、功能最广泛的蛋白家族,其可能涉及植物次生代谢物的跨膜转运,从而保护植物免受环境胁迫的伤害[34-36]。这些参与早期特异性途径的基因在榆叶梅早期碱性响应机制中可能起着重要的作用,并可能在后续植物响应碱胁迫中诱导产生级联反应。本研究中,在碱胁迫处理6、12 h 条件下,开始涉及许多重要代谢途径,包括淀粉和蔗糖代谢途径、植物激素信号转导途径、糖酵解/糖异生途径、光合作用途径、类胡萝卜素生物合成途径,以及光合作用-天线蛋白途径等,表明这些参与代谢途径的基因随着碱胁迫时间的增加而呈多样性表达模式,并且榆叶梅在碱胁迫适应过程中存在快速、多样的信号感知、转导以及相应复杂的分子机制。

图5 3 h vs.0 h、6 h vs.0 h 和12 h vs.0 h 的GO 生物学过程类型对比图Fig.5 Comparative distribution of GO enrichment of terms related to biological processes for 3 h vs.0 h, 6 h vs.0 h and 12 h vs.0 h
本试验结果表明,RT-qPCR 所得基因的表达模式与RNA-Seq 数据结果类似,但它们间仍存在一些差异,这可能是RNA-Seq 和RT-qPCR 两种方法使用的算法和原理不同而致[37-38],这与Yates 等[39]和Huang等[40]的研究结果类似。此外,这7 个基因在3 种碱胁迫时间处理下均呈极显著性差异表达(P<0.01),表明它们可能在响应碱胁迫过程中发挥作用。总之,本研究从RT-qPCR 和RNA-Seq 方法中获得了类似的基因表达模式,证实了测序结果的可靠性和准确性。对于重要的差异表达基因,后续还将进一步用northern blot对其进行验证。
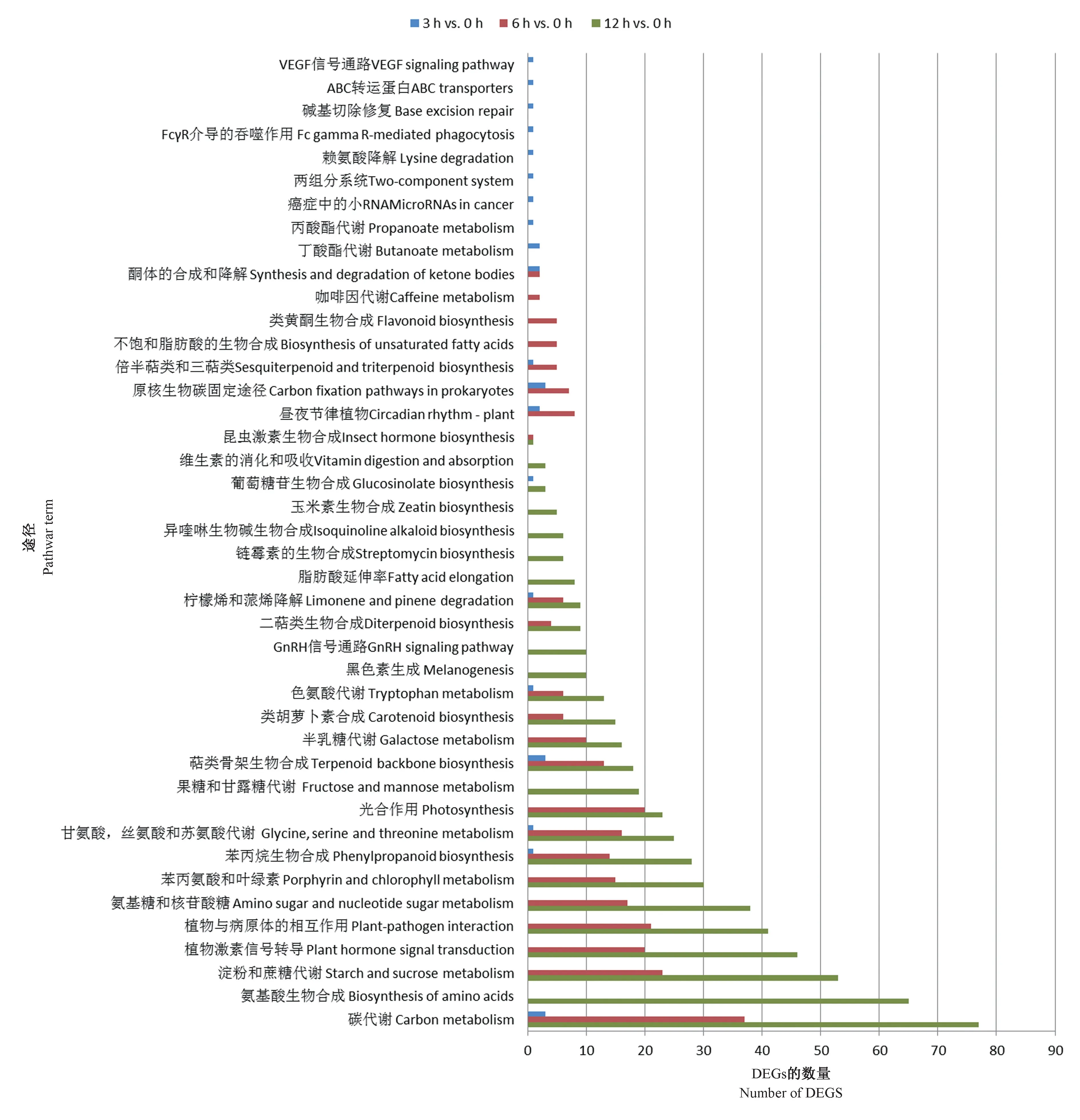
图6 3 h vs.0 h、6 h vs.0 h 和12 h vs.0 h DEGs 的KEGG 富集分析Fig.6 KEGG enrichment analysis of the DEGs of 3 h vs.0 h, 6 h vs.0 h and 12 h vs.0 h
叶片作为植物进行光合作用和生命活动的主要器官,是对逆境刺激反应最为敏感的部位。本研究主要通过叶片的分子变化来探索榆叶梅应答碱胁迫的机制。目前,在蔷薇科果树上有研究报道[41],水分胁迫下,新根内皮层细胞出现细胞壁增厚现象,对于榆叶梅根系在碱胁迫下的响应机制将进行后续研究。本研究发现了一些碱胁迫下差异表达的基因,可能对应调控不同的物质或途径,为将来采用分子生物学的手段将单个抗性基因导入植株体,获得抗碱转基因材料、培育耐碱果树新品种创造前提条件。此外,本研究是针对榆叶梅砧木本身进行碱胁迫响应机制的分析,而嫁接苗的抗性机制与砧木又不完全相同,后续会以榆叶梅为砧木嫁接不同李品种,并与毛桃砧木嫁接苗对比研究,进一步探讨砧木对李抗碱性的影响及其内在机理。
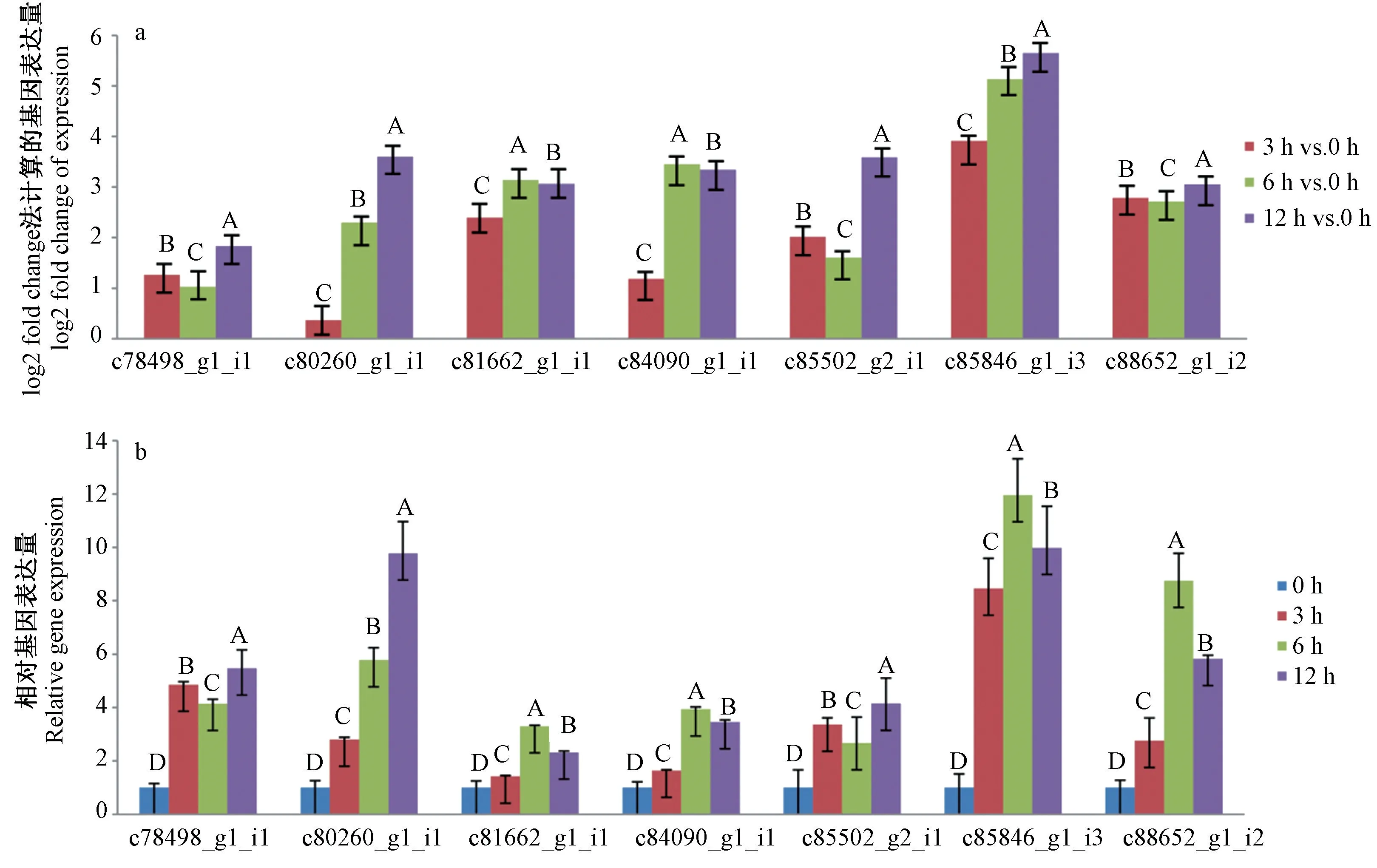
图7 榆叶梅7 个差异表达耐碱基因的RNA-Seq 和RT-qPCR 表达模式图Fig.7 Expression patterns of seven genes selected from alkalinity-responsive DEGs of Prunus triloba by RNA-Seq and RT-qPCR
4 结论
本研究对榆叶梅在单一碱胁迫下的分子响应机制进行了探索。使用Illumina 测序平台对榆叶梅在短期碱胁迫下的叶片进行了de novo转录组分析,获得了大量的DEGs,可用于榆叶梅今后的分子育种中。基于这些DEGs,进行GO 富集分析发现,其中28 个基因可能在早期碱胁迫响应机制中起着重要的作用;而对这些DEGs 进行KEGG 富集分析发现,不同碱处理时间下的KEGG 途径具有显著差异。此外,采用RT-qPCR 法验证了7 个碱性相关基因的表达模式,证实了RNASeq 结果的可靠性。本研究获得了关于榆叶梅对碱胁迫的短期分子响应机制的相关数据,可为了解榆叶梅及其他相关果树的短期碱胁迫分子适应机制奠定基础。
