聚乳酸/聚丁二酸丁二酯扩链改性材料研究
2020-06-22刘洒文高山俊沈春晖齐宏生陈秀玲
刘洒文,高山俊,沈春晖,齐宏生,陈秀玲
(武汉理工大学材料科学与工程学院,武汉 430070)
聚乳酸(PLA)具有高拉伸强度以及良好的生物降解性能,不仅在使用过程中不会对环境造成污染,其单体组成乳酸也属于生物可再生资源,可由玉米或淀粉发酵得到。此外,由于对人体无害,且具有良好的生物相容性,被广泛应用于生物医疗材料领域。作为高分子材料,较高的分子量是PLA 获得应用的重要前提。目前研究者已通过多种方法制备了较高分子量PLA,如熔融/固相缩聚法、溶液缩聚法、扩链法等。现阶段主要合成方法是丙交酯开环聚合法,该法制得的PLA 分子量高,分子量分布较窄,聚合时间较短,但缺点是制备工艺复杂,聚合条件苛刻,生产成本高。此外,PLA 还具有脆性易断的特点,这都是制约PLA 应用的问题所在[1]。
聚丁二酸丁二酯(PBS)也是有生物降解性的脂肪族聚酯,具备良好的热稳定性,力学性能与聚乙烯和聚丙烯类似。单体组成丁二酸来源广泛且价格低廉,可从玉米、乳清等农业产品中获得,最为重要的是PBS 链段为柔性链,具有一定的韧性,可以改善PLA 的脆性特点[2]。
生产制备高分子材料过程中,共混是使材料性能复合化的有效方法之一,但是由于大部分聚合物间的相容性很差,造成共混过程中连续相与分散相间界面粘合力差,最终导致低劣的材料性能。单纯将PLA 与PBS 物理共混的增韧效果极差,脆性基本得不到改善,故考虑使用扩链剂原位增容,以期改善性能。
笔者使用扩链剂4,4′–二苯基甲烷二异氰酸酯(MDI)和 2,2′–(1,3– 亚苯基 )– 二恶唑啉 (PBO),通过与左旋PLA(PLLA)和PBS 的端羟基及端羧基反应,在熔融条件下制备了PLLA/PBS 扩链改性材料。其中PLLA 通过左旋乳酸(L–LA)直接缩合得到,虽然分子量比开环聚合PLLA 要小,但是共混体系中PLLA 形成的脆性区域也相应减少,较低的分子量通过两步扩链反而得到弥补甚至大大提高,不仅避开了合成PLA 传统开环聚合方法的弊端,还提升了材料韧性及其它力学性能,达到实验目的[3]。
1 实验部分
1.1 主要原料
L–LA :CAS 号 79–33–4,纯度 80%,湖北健文生物医药有限公司;
PBS:工业级,安庆和兴化工有限公司;
MDI:CAS 号 101–68–1,阿拉丁生化科技股份有限公司;
PBO:郑州华盛化工有限公司;
辛酸亚锡:CAS 号 301–10–0,纯度 95%,阿拉丁生化科技股份有限公司;
对甲苯磺酸:CAS 号 104–150–4,分析纯,湖北鑫润德化工有限公司;
二氯甲烷:CAS 号 75–09–2,二氯甲烷,99.5%,阿拉丁生化科技股份有限公司。
1.2 主要设备和仪器
电子天平:TMP–2 型,湘仪天平仪器设备有限公司;
鼓风干燥箱:DHG–9070A 型,上海索域实验设备公司;
密炼机:SU–70 型,常州苏研科技有限公司;
热压机:R–3201 型,武汉启恩科技发展有限公司;
注塑机:M1200 型,武汉启恩科技发展有限公司;
万能试验机:CMT4104 型,美特斯工业系统有限公司;
熔融指数仪:ZRZ–400 型,新三思材料检测公司;
傅里叶变换红外光谱仪(FTIR):EQUINOX–55型,德国布鲁克公司;
核磁共振仪: AVANCE Ⅲ 500 MHz 型,德国布鲁克公司;
场发射扫描电子显微镜(SEM):Zeiss Ultra Plus 型,德国蔡司光学仪器公司;
综合热分析仪:STA449F3 型,德国耐驰机械仪器有限公司;
冲击性能测试仪:RESIL–6957 型,意大利Ceast 公司。
1.3 样品制备
(1) PLLA 的合成。
将150 g 的L–LA 加入到三口烧瓶中,充氮气且在一定真空度条件下通过油浴锅加热至50℃,直至不再蒸出液体为止。再以2℃/min 速率缓慢上升至100℃,维持30 min 提纯处理,最终得到无色透明的L–LA。
将除杂处理后的100 g L–LA,0.4%辛酸亚锡(占L–LA 的质量比,下同)和0.4%对甲苯磺酸组成的复合催化剂加入到三口烧瓶中。在磁力搅拌、氮气保护及一定真空度的条件下,通过油浴锅升温至150℃,此后进一步搅拌脱水4 h,然后以2℃/min 的速率升温至180℃连续反应8 h,停止反应并取出产物。将反应得到的乳酸低聚物溶于二氯甲烷中,搅拌使其完全溶解,再倒入进行过冷却处理的无水甲醇中,用玻璃棒搅拌析出白色的乳酸低聚物,放入60℃的真空烘箱中干燥24 h 备用。
将乳酸低聚物,0.4%辛酸亚锡与0.4%对甲苯磺酸组成的复合催化剂加入到250 mL 三口烧瓶中。在机械搅拌、氮气保护及一定真空度的条件下,通过油浴加热至105℃热处理2 h,再升温至150℃固相反应20 h 出料,待反应产物冷却至室温后得到褐色产物。将反应产物溶于二氯甲烷中,搅拌使其完全溶解,再倒入进行过冷却处理的无水甲醇中,用玻璃棒搅拌析出PLLA,放入60℃的真空烘箱中干燥 24 h 备用[4]。
(2) PLLA/PBS 扩链改性料制备。
将PLLA 和PBS 分别放在鼓风干燥箱中80℃干燥12 h,称取PLLA 与PBS 组分质量比为3/1,混合均匀后加入密炼机搅拌10 min,温度设定为185℃,转速为 55 r/min,加入一定量的 PBO 扩链剂熔融反应15 min,扩链反应式见图1。降温取出密炼料,用热压机压片后剪条切粒,干燥处理后注塑成型力学性能测试样条。相关参数设置为:注塑温度170℃,注塑时间12 s,合模时间5 s,冷却时间6 s,注塑压力6 MPa,模具在使用前放入鼓风干燥箱50℃预热1 h[5]。确定PBO 含量后,称取一定量的MDI,以相同方法制样,扩链反应式见图2。不同试样配方见表1。

图1 扩链增容第一步反应式
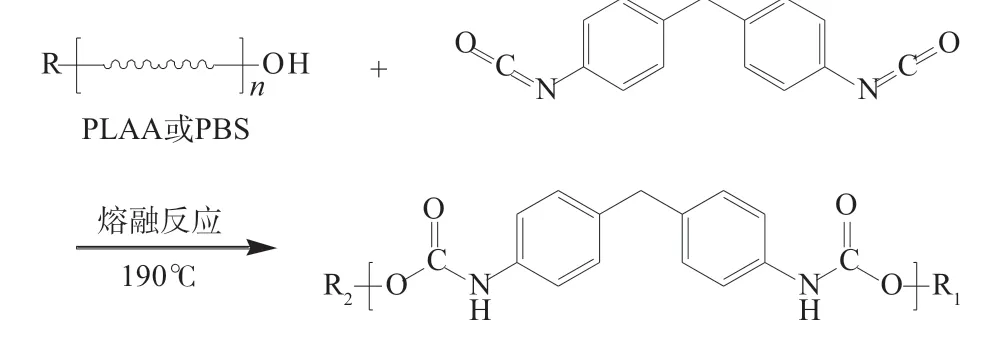
图2 扩链增容第二步反应式
1.4 性能测试和结构表征
酸值通过滴定法测定,定义为中和1 g 聚合物所需的KOH 的量,将样品溶解于氯仿中,并在萘酚酞指示剂存在下,在乙醇溶液中用0.01 mol/LKOH 滴定,计算端羧基数量;
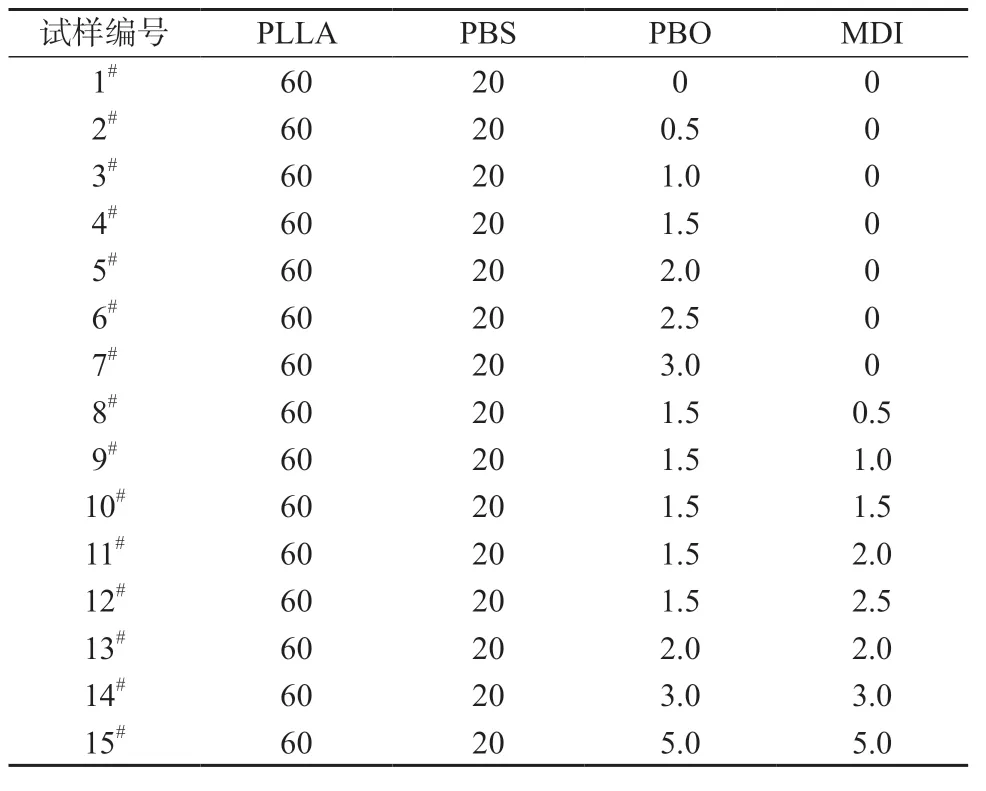
表1 试样配方 g
核磁共振谱(1H–NMR)分析:以氘代氯仿为溶剂,四甲基硅烷(TMS)为内标。
FTIR 分 析:采 用 KBr 压 片 制 样,于 400~4 000 cm–1范围内制得红外谱图。
差示扫描量热(DSC)分析:在氮气气氛下,快速升温至140℃保持5 min 消除热历史,再以10℃/min 速率降温到10℃,保持3 min,然后以10℃/min 速率升温至190℃,在此范围内记录曲线,并计算结晶度。
热重分析(TGA)测试:氮气保护,升温速率10℃/min,温度范围 20~500℃。
断面形貌分析:将测试材料制成10 mm×50 mm 的长方体样条,液氮冷冻处理后对断面进行喷金处理,接着用SEM 冷场模式观察断面形貌。
拉伸性能:使用万能试验机按照GB/T 528–2009 测试,室温,拉伸速率50 mm/min,哑铃状样条,尺寸75 mm×5 mm×2 mm,每组测试5 根样条取平均值。
悬臂梁冲击强度:按GB/T 1843–1996 测试,样条宽6 mm、厚4 mm,每组测试5 根样条取平均值。
MFR 测试:温 度 155 ℃,预热 3 min,负重2 160 g,切割间隔10 s,每次试样切割10 次,排除极大极小值后取平均值[6–7]。
2 结果与讨论
2.1 酸值滴定法分析
PBO 与羧基的反应活性很高,而对脂肪醇呈惰性,因此可以选择性地改性聚酯的羧基端基,没有明显的副反应。MDI 也可以与羧基端基反应生成酰胺、羧酸酐或脲,但反应速度不够快,为了避免该反应,选择先加入PBO 与 PLLA/PBS 反应,以减少共混体系的酸值,保留较多的羟基来完成第二步与MDI 进行反应。封端反应和偶联反应都降低了第一步扩链反应产物的羧基含量,这对MDI 链接反应是十分有利的,在第一步15 min 的熔融共混反应过程中,产物的酸值从11 mg KOH/g 降至3 mg KOH/g,这个数值属于典型的羟基封端的聚合物水平[18]。若第一步选择MDI 与PLLA/PBS 反应,在15 min 的反应后,样品的酸值从11 mg KOH/g 降至9 mg KOH/g,反应时间增至1 h 后,酸值也只降至 7 mg KOH/g[3]。
2.2 结构表征测试
图3 为各阶段产物的1H–NMR 谱图。其中氘代氯仿的溶剂峰的化学位移δ为7.26,TMS 内标峰δ为0,PLA 结构中甲基基团和次甲基基团的氢峰δ值分别是 1.56(1),5.24(2),两峰的积分值比约为3 ∶ 1,符合 PLLA 的谱图特征[1]。在 PBS 的谱图中,重复单元中不同亚甲基的氢峰δ值为1.69(3),2.61(5)和4.10(4),从峰面积计算各氢原子个数比例为1 ∶1 ∶1,总体符合PBS 的谱图特征[1]。

图3 各阶段产物的1H–NMR 谱图
PLLA–PBO–PBS 的谱图中,PLLA 与 PBS 基团的氢峰在图中(1~5 位置)能明显观测到,且从峰面积计算的各氢原子个数比例符合PLLA/PBS配比 3/1 的比例,羟基基团的氢峰δ为 2.17(6),酰胺基团的氢峰的δ值则出现在1.25(7),共聚物苯环基团的峰δ值在7.48(8),各峰值所代表的结构与预测的目标产物结构一致。
PLLA–PBO–MDI–PBS 的谱图中,苯环上的氢峰δ值为 7.48(4),次氨基的氢峰的δ值为 1.25(7),而原本的羟基峰(6)与MDI 反应,生成酰胺键而消失,各峰值所代表结构与预测目标产物一致。
图 4 为产物的 FTIR 谱图。PLLA–PBO–PBS 的谱图中,3 503 cm–1左右处为酯羟基—OH 的对称伸缩振动峰,2 995 cm–1和 2 946 cm–1处为甲基以及亚甲基的伸缩振动峰;1 759 cm–1处为酯羰基的对称伸缩振动峰,酰胺基的特征振动峰在1 689 cm–1(酰胺 I 带 )、1 531 cm–1(酰胺Ⅱ带 )、1 290 cm–1(酰胺Ⅲ带)处出现且会在3 540 cm–1附近出现两个尖的吸收带,此外1 657 cm–1处还出现苯环骨架特殊吸收峰[8],以上特征吸收峰都说明得到了目标产物PLLA–PBO–PBS。

图4 各阶段产物的FTIR 谱图
在 PLLA–PBO–MDI–PBS 的 FTIR 谱 图 中,1 532 cm–1,3 376 cm–1峰 出 现,和 PLLA/PBO/PBS 的谱图对比,吸收峰面积明显增大,这是聚氨酯 N—H 键的特征吸收峰;1 689 cm–1(酰胺 I 带 )、1 531 cm–1(酰胺Ⅱ带 )以及 3 540 cm–1附近常出现两个尖的吸收带的峰面积也都增大;1 598 cm–1,1 456 cm–1属于苯环骨架的特征吸收峰,也出现明显扩大,这都表明了初产物与MDI 进一步发生扩链反应[9]。
2.3 热力学性能测试
图5 为扩链产物的DSC 曲线,相应数据见表2,其中Tg为玻璃化转变温度,Tc为冷结晶温度,Tm1为 PBS 的熔点,Tm2为 PLA 的熔点,Xc为结晶度。可以看出PBO 和MDI 扩链增容剂都有提高产物热稳定性的效果,经扩链共混后,产物的Tg与Tc出现上升的趋势,这是因为通过扩链改性,使产物的分子量迅速提高,同时两步增容反应都引入了苯环,使得分子链的刚性增强,分子链想达到运动的程度需要更多的能量,所以增加了热稳定性。而两相的熔点变化不大,只是熔融峰面积出现明显的减小趋势,这是因为苯环的加入、分子量的增加以及酰胺基的生成都会使分子链难以规整排列,极大地增加了结晶的难度,Xc急剧减小,这意味着改性后的产物具有更好的韧性和延展性,符合改善PLLA 脆性的实验目的[10]。另外,共混物的Tg也更加靠近文献记载的纯PLLA 和纯PBS 的Tg的中间值,意味着实现了产物相容性提高的目的,这主要就是因为扩链剂PBO 与MDI 和PLLA/PBS 端基的化学链接作用,属于原位反应增容[11]。

图5 扩链产物DSC 曲线

表2 扩链产物DSC 曲线数据

图6 扩链产物TG 和DTG 曲线

表3 扩链产物热力学参数
图6 为扩链产物的TG 和DTG 曲线,相关参数见表3,其中T5%为分解时的温度,Tmax为最大分解速率温度,Dmax为最大分解速率。通过对比可以发现,经过 PBO 和 MDI 扩链的产物的T5%,Tmax都出现显著提高,并且Dmax也有所下降,可以很明显看到在进一步使用MDI 后或增加扩链增容剂用量后,材料的热稳定性显著提高。这是因为通过增容扩链,材料的分子量有了跨越式增长,而且PBO 与MDI小分子中都含有苯环基团,和通过扩链反应后形成的酰胺基一样,都是热稳定性更高的基团。这些因素的共同作用才使得材料的热稳定性显著提高[12]。
2.4 SEM 分析
图7 为不同阶段产物的SEM 照片。从图7 可以看出,PLLA/PBS 共混体系相容性不好,两相的区分明显;加入PBO 后相界面虽然还存在缺陷和空隙,但是相容性已经大大提升;而在加入MDI 熔融共混后,断面更加均匀致密,扩链剂加入量相对更多时,基本已经是各向同性的物质了,不能充分发挥共混物两相的优点,所以在力学测试中,反而具有更高的脆性特征[13]。

图7 扩链产物SEM 照片
2.5 力学性能分析
PLLA/PBS 基复合材料的拉伸性能见图8。纯PBS 的拉伸强度为34.4 MPa,而纯PLLA 的拉伸强度可以达到69.1 MPa。可以观察到,不加入扩链剂的PLLA/PBS 共混物的强度约为30 MPa,当加入1.8% (1.5 g)的PBO 后,扩链剂与羧基的反应使两相的相容性得到提升,PLLA 和PBS 各自的优点得以体现在共混体系中,材料的拉伸强度得到了较大提升,增加到52.6 MPa,而当进一步提高PBO的加入量后,拉伸强度略有下降,原因推测为两相体系经过扩链反应相连后,共混物相容性突破临界度,整个体系的性质趋向完全相容,不能结合体现PLLA 的拉伸强度优点和PBS 的韧性,且未反应的PBO 也将影响微观结构的结晶过程,使分子链排列杂乱,Xc和拉伸强度出现下降。

图8 复合材料的拉伸性能
确定PBO 最佳配比后再加入MDI 使体系内羟基反应扩链,增加分子量。可以观察到,当加入1.8% (1.5 g)的MDI 后,复合材料的拉伸强度进一步提高,增加至72 MPa。而当加大投入量后也出现拉伸强度下降的趋势,原因推测为更大的分子量使得分子链运动困难,有效反应减少。而加入另一种扩链剂MDI 能进一步提高材料拉伸强度的原因是扩链反应的连接点不同,化学反应不同,适当配比的MDI 加入使已经过PBO 扩链反应的产物分子量大幅上升,且新的酰胺键生成更进一步增大了材料的拉伸强度。PLLA/PBS 共混物断裂伸长率仅有5%左右,且材料脆性明显,当加入约1.8%的PBO后,材料的断裂伸长率增加到75.6%,原因是扩链剂PBO 有效增加了PLLA 和PBO 相容性,而当取最佳配比的PLLA–PBO–PBS 的产物再与1.8%的MDI 熔融共混后,网状的链纠缠结构以及高分子量使材料的断裂伸长率达到129.4%。
图9 示出了材料的冲击强度变化。可以看出当只添加了PBO 增容时,强度只是略有提升,显著变化发生在第二步反应,推测是新的扩链反应链接点使微观结构连接更紧密,通过分子链纠缠,更有助于对抗外加冲击。

图9 复合材料的冲击强度
2.6 MFR 测试
对于PBS 而言,MFR 过高、流动性太好而难以工业加工是其缺点之一,而PLLA 的MFR 则处于合适的加工范围。实验研究发现,PLLA/PBS 共混体系的MFR 会随PBS 加入量的变化而发生较大变化[14],最佳配比 3 ∶ 1 时,MFR 为 25.64 g/10 min,而配比为 1 ∶ 1 时,MFR 达到了 69 g/10 min。实验在改善PLLA/PBS 共混物脆性特征的同时,也希望能使材料具有更合适的流动性以便于加工。图10 为材料不同配比时的MFR。随着扩链增容剂的添加,产物的MFR 呈下降趋势,总体符合添加量越多,MFR 越低的反比关系,但存在临界值。出现这个结果的原因除了扩链剂让共混体系分子量大幅上升外,也有扩链剂中苯环等大基团的阻隔作用,使原本的脂肪族高分子单链结构拓展为互相交联的网状结构,极大增加了分子运动的困难度。而当体系中端羟基、端羧基扩链反应点反应完全后,未反应的扩链添加剂作为小分子存在体系中,增大了分子链间隙,反而使MFR 有了少许提升[15–16]。

图10 不同配比材料的MFR
3 结论
使用 L–LA 合成 PLLA,先用 PBO 扩链剂与PLLA/PBS 发生反应,再加入MDI 进一步反应,通过熔融反应制得产物。在扩链反应时,PBO 加入量为1.8%且MDI 加入量为1.8%时,扩链反应效果最好,材料能获得最佳的韧性和拉伸强度。韧性的提高一是通过苯环的加入,从而降低材料的结晶度,二是羧基及羟基与扩链剂的反应,生成不同种类的化学链接和网状结构,并同时提高了拉伸强度。同时,两种不同类型且相互促进的扩链剂使PLLA 直接缩合成为可能,避开合成PLA 传统的开环聚合方法制备工艺复杂、聚合条件苛刻、生产成本较高的缺点。最终实验产品不仅提高了热稳定性,改善了加工性,还使拉伸强度提高了140%,材料发生明显的脆性–韧性转变,基本实现了最初实验目的。
