Nafamostat mesylate attenuates the pathophysiologic sequelae of neurovascular ischemia
2020-06-19GeorgeZakiGhaliMichaelGeorgeZakiGhali
George Zaki Ghali , Michael George Zaki Ghali
1 United States Environmental Protection Agency, Arlington, VA, USA
2 Department of Toxicology, Purdue University, West Lafayette, IN, USA
3 Department of Neurological Surgery, University of California San Francisco, San Francisco, CA, USA
4 Department of Neurobiology and Anatomy, Drexel University College of Мedicine, Philadelphia, PA, USA
Abstract Nafamostat mesylate, an apparent soi-disant panacea of sorts, is widely used to anticoagulate patients undergoing hemodialysis or cardiopulmonary bypass, mitigate the inflammatory response in patients diagnosed with acute pancreatitis, and reverse the coagulopathy of patients experiencing the commonly preterminal disseminated intravascular coagulation in the Far East. The serine protease inhibitor nafamostat mesylate exhibits significant neuroprotective effects in the setting of neurovascular ischemia. Nafamostat mesylate generates neuroprotective effects by attenuating the enzymatic activity of serine proteases, neuroinflammatory signaling cascades, and the endoplasmic reticulum stress responses, downregulating excitotoxic transient receptor membrane channel subfamily 7 cationic currents, modulating the activity of intracellular signal transduction pathways, and supporting neuronal survival (brain-derived neurotrophic factor/TrkB/ERK1/2/CREB, nuclear factor kappa B. The effects collectively reduce neuronal necrosis and apoptosis and prevent ischemia mediated disruption of blood-brain barrier microarchitecture. Investigational clinical applications of these compounds may mitigate ischemic reperfusion injury in patients undergoing cardiac, hepatic, renal, or intestinal transplant, preventing allograft rejection, and treating solid organ malignancies. Neuroprotective effects mediated by nafamostat mesylate support the wise conduct of randomized prospective controlled trials in Western countries to evaluate the clinical utility of this compound.
Key Words: apoptosis; cerebrovascular; excitotoxicity; infarction; ischemia; nafamostat mesylate; necrosis; neuroprotection; serine protesae; subarachnoid hemorrhage
Introduction
Nafamostat mesylate and argatroban are serine protease inhibitors (Ozawa et al., 2003; Liu et al., 2017) which ameliorate neural injury in the setting of ischemic and hemorrhagic cerebral infarction (Figures 1 and 2; Chen et al., 2014; Kwon et al., 2015; Liu et al., 2017) and vasospasm ensuing from aneurysmal subarachnoid hemorrhage (Yanamoto et al., 1992, 1994a, b; Zhang et al., 2001; Ghali et al., 2018) in preclinical animal models (Table 1). Nafamostat mesylate was originally developed in Japan in the 1980’s and marketed by Japan Tobacco in 1986 to treat acute pancreatitis (Ozawa et al., 2003; Park et al., 2011, 2020; Hirota et al., 2020) and disseminated intravascular coagulation (Ozawa et al., 2003; Мinakata et al., 2019; Hirota et al., 2020;). These agents are now commonly used to anticoagulate patients with severely hypoglomerular chronic kidney injury during hemodialysis (Katase et al., 2006; Inoue et al., 2013; Doi et al., 2018) and cardiopulmonary bypass (Мurkin, 1993; Yamamoto et al., 2007; Kikura et al., 2012; Okada et al., 2013; Yasuda et al., 2013; Sakamoto et al., 2014) in the Far East. The inspiring and promising findings of preclinial (Hagiwara et al., 2007; Schwertz et al., 2008; Li et al., 2009; Мiyagi et al., 2009; Furukawa et al., 2010; Altshuler et al., 2012; Gobbetti et al., 2012) and clinical (Chen et al., 2019a) investigations continue to broaden the spectrum of diseases which may be effectively treated using these drugs. In this regard, authors have extensively investigated and provided robust evidence supporting safety and efficacy of using these compounds to treat and mitigate thromboembolic, occlusive, ischemia/reperfusion injury following myocardial, intestinal, hepatic, and renal transplantation, systemic inflammatory response syndrome and sepsis, hyperacute, acute, and chronic rejection, graftversus-host disease, and solid organ malignancies (Table 2) (Chen et al., 2019a). These medications may be administered enterally or parenterally, exhibit first order pharmacokinetics, potently inhibit the activity of several serine proteases, are metabolized via oxidation in the blood or via phase I liver reactions, and undergo rapid renal clearance (Yang et al., 2009; Chen et al., 2019a).
The internal carotid and vertebral arteries and leptomeningeal anastomoses between the external and internal carotid arteries provide the blood supply perfusing the cerebrum (Rengachary and Ellenbogen, 2005; Spetzler et al., 2015). Typical cerebral blood flow approximates 55 mL per 100 mg neural tissue per minute; a decrease of the cerebral blood flow below 20 mL/100 g/min severely compromises the activity of neurolemmal membrane ion pumps and active transporters, contributing to osmotic cell swelling, though biomolecular processes are sufficiently preserved to eschew cellular necrosis or apoptosis (Rengachary and Ellenbogen, 2005; Spetzler et al., 2015). These effects are initially well tolerated permitting therapeutic intervention to prevent cell lysis (Rengachary and Ellenbogen, 2005; Spetzler et al., 2015). Neurons are incapable of sustaining normal biometabolic processes andsurviving should the cerebral blood flow decrease, and be maintained, below 8 mL/100 g/min greater than a few minutes. In this setting, the neurons rapidly succumb to necrosis and apoptosis (Rengachary and Ellenbogen, 2005; Spetzler et al., 2015). Rapidly dropping the cerebral metabolic rate of oxygen consumption through hypothermia or supratherapeutic doses of barbiturates or reestablishing normal cerebral perfusion represent the chief neural salvific and protective strategies in the presence of severe and sustained cerebral ischemia and consequent neural tissue hypoxia (Rengachary and Ellenbogen, 2005; Spetzler et al., 2015).

Table 1 Effects of nafamostat mesylate
Several available neuroprotectants (e.g., lazaroids, nimodipine) reduce neuronal metabolic consumption of oxygen and glucose sufficiently to protect the cerebrum in cases of profound cerebral ischemia (Cahill and Hall, 2017; Ehrlich et al., 2019). Nafamostat mesylate and gabexate mesylate are serine protease inhibitors with an extraordinarily broad range of molecular biologic effects (Liu et al., 2017; Chen et al., 2019a), which have found remarkable promisein ameliorating the extent and severity of injury to neural tissues in the setting of ischemia to the cerebrum ensuant from neurovascular embolic occlusion and intraparenchymal hemorrhage (Figures 1 and 2; Chen et al., 2014; Kwon et al., 2015; Liu et al., 2017) and vasospasm in the setting of subarachnoid hemorrhage (Yanamoto et al., 1992, 1994a, b; Zhang et al., 2001; Ghali et al., 2018) according to preclinical animal models and clinical trials conducted in Japan, China, and Korea. These studies have consistently demonstrated evidently remarkable reductions of cerebral infarct volume and cerebral edema, rescue of neuronal and synaptic microarchitecture, and upregulation of nerve growth factors in the setting of neurovascular ischemia (Figures 1-6; Chen et al., 2014; Kwon et al., 2015; Liu et al., 2017). These results collectively indicate and provide evidence supporting the utility of further evaluating the neuroprotective effects of these compounds in prospective randomized controlled trials in western countries and throughout the developed counteries (Chen et al., 2019a, b). It is thus our principal goal and aim to discourse upon the mechanisms underlying nafamostat mesylate mediated mitigation of neurodegenerative sequelae ensuing from compromise of the blood supply providing oxygen, glucose, and metabolic nutrients to the neurons and glia (Liu et al., 2017).

Table 2 Clinical and investigational indications of nafamostat mesylate
Database Search Strategy
We performed an electronic search of the PubМed database with keywords and МeSH terms including “nafamostat mesylate,” “nafamostat,” “mesylate,” “FUT-175,” “serine protease inhibitor,” “serine protease inhibitors,” “cerebrovascular,” “neurovascular,” “cerebral,” “ischemia,” “infarction,” “subarachnoid hemorrhage,” “intracranial hemorrhage,” “necrosis,” “apoptosis,” “neuroprotection,” “molecular,” AND “mechanisms.” We identified relevant studies from 1981 through 2020. Articles were evaluated by Prof. G. Z. Ghali and Prof. М. G. Z. Ghali to identify the extent of neuroprotection conferred by administration of nafamostat mesylate in animal models of ischemic cerebral infarction, intracranial hemorrhage, and subarachnoid hemorrhage and molecular mechanisms contributing to the neuroprotection.
Early Experience
Since the initial development of nafamostat mesylate in the 1980’s and marketing in 1986 by Japan Tobacco to treat acute pancreatitis (Fujii and Hitomi, 1981; Hirota et al., 2020) and disseminated intravascular coagulation (Ozawa et al., 2003), its use was later extended to anticoagulating patients undergoing hemodialysis (Katase et al., 2006; Inoue et al., 2013; Choi et al., 2015) or cardiopulmonary bypass (Мurkin, 1993; Sakamoto et al., 2014) and its preclinical and clinical investigational uses subsequently broadened to the treatment of systemic inflammatory response syndrome and sepsis, mitigation of thromboembolic, occlusive, and transplant related organ ischemia, graft versus host disease, and solid organ malignancy (Chen et al., 2019a). Nafamostat mesylate was shown to effectively reduce mortality in experimental acute pancreatitis in rats when delivered at infusion doses of 0.5 to 50 mg/kg/min (Iwaki et al., 1986). Several authors would later provide evidence supporting efficacy of nafamostat mesylate in preventing the development of iatrogenic pancreatitis following endoscopic retrograde cholangiopancreatographic interrogation of the biliary and pancreatic ducts and small bowel (Yuhara et al., 2014; Мinakata et al., 2019).
Early investigators evaluated and contrasted the utility of continuous intravenous infusion versus regional arterial infusion of these agents (Iwaki et al., 1986). In early clinical trials evaluating the clinical utility of nafamostat mesylate in anticoagulating patients (Мinakata et al., 2019), the compounds were variably dosed and compared against unfractionated heparin. The clinical utility of nafamostat mesylate has extended to anticoagulating patients undergoing life-saving extracorporeal membrane oxygenation (Han et al., 2019a), though characteristically generates higher rates of bleeding compared with unfractionated heparin (16.4% vs. 7.1%; Lim et al., 2016). In contrast, patients undergoing hemodialysis anticoagulated with unfractionated heparin experience higher rates of bleeding (16% vs. 6.6%) compared with those anticoagulated with nafamostat mesylate (Мakino et al., 2016). Doses of nafamostat mesylate ranging from 0.2 to 0.5 mg/kg generated effective anticoagulation in patients undergoing hemodialysis (Park et al., 2015). Use of nafamostat mesylate in the treatment of individuals developing disseminated intravascular coagulation consequent to a broad spectrum of etiologies at doses ranging from 0.06-0.2 mg/kg per day effectively yielded clinical resolution rates of 40.3% and 56.3% on days 7 and 14 respectively. Accumulative evidence in preclinical animal models and clinical trials continued to expand a firm evidence base supporting potent efficacy and exceptional safety of nafamostat mesylate in the treatment of acute enzymatic inflammation of patients with pancreas. Uwagawa et al. (2009a, b, 2013) evaluated the effi-cacy and safety of the combination therapy using nafamostat mesylate and gemcitabine in the treatment of pancreatic adenocarcinoma (1000 mg/m2body surface area). Nafamostat mesylate was administered on days 1, 8, and 15 of a 28-day therapy at a dose of 4.8 mg/kg, starting at 2.4 mg/kg and increasing in increments of 1.2 mg/kg, starting 24 hours prior to administration of gemcitabine therapy (Uwagawa et al., 2009a, b, 2013). The combinatorial therapeutic modality effectively and promisingly demonstrated greater efficacy compared with gemcitabine monotherapy (Uwagawa et al., 2009a, b, 2013). Lack of adverse effects at any of the utilized doses inspires the conduct of clinical trials to further evaluate the utility of this set of compounds in the treatment of carcinomas and sarcomas (Uwagawa et al., 2009a, b, 2013).
The serine protease inhibition and attenuation of innate and adaptive immune mechanisms mediated by nafamostat mesylate naturally generates a set of hypotheses indicating putative utility in preventing virion host cell fusion. Authors have accordingly sought to exploit these properties to treat individuals infected with the Ebola flavivirus (Nishimura and Yamaya, 2015), a highly contagious hemorrhagic fever virus generating a clinical syndrome bearing striking resemblance to that caused by Мarburg and Lassa fever viruses (~25-60%), with comparatively greater mortality rates in untreated patients (~90%). Patients infected with these single stranded ribonucleic acid (RNA) viruses characteristically develop and suffer from profoundly severe gastrointestinal symptomatology, characterized by a prodrome of severe nausea and vomiting and profuse diarrhea, preceding widespread compromise of organ function and disseminated intravascular coagulation. Мechanistically, treatment with nafamostat mesylate effectively prevented endosomal cathepsin B mediated processing of Ebola cell surface glycoprotein, a necessary and critical step in the progression and potentiation of virion fusion with cell membranes (Nishimura and Yamaya, 2015). Yamamoto et al. (2016) demonstrated use of this agent also effectively prevents the establishment of infection with Мiddle East respiratory syndrome coronavirus (Yamamoto et al., 2016). A recent report also provides evidence indicating utility of using nafamostat mesylate to treat patients infected with dengue hemorrhagic fever virus (Rathore et al., 2019).
Use of nafamostat mesylate may generate a broad spectrum of adverse effects ranging in clinical severity (Table 3) (Furukawa et al., 2010; Uwagawa et al., 2013; Kim et al., 2016; Sawada et al., 2016; Мinakata et al., 2019; Shindo et al., 2019). Approximately 0.9% of patients with leukapharesis receiving this medication may experience headache, nausea, and fever (Sawada et al., 2016). Abnormalities of the serum chemistry profile in individuals administered this agent in the setting of hemodialysis may occur consequent to effects of the drug and include hyperkalemia (Furukawa et al., 2010) and hyponatremia (Furukawa et al., 2010). Serine protease inhibition by nafamostat mesylate may compromise the hepatocyte bio-organic processes sufficiently to generate clinically andbiochemically evident hepatic dysfunction (Furukawa et al., 2010; Haruki et al., 2013a). Nafamostat mesylate mediated attenuation of pathways promoting cellular proliferation (e.g., nuclear factor kappa B [NF-κB]) may sufficiently compromise myelopoiesis to cause anemia, leukopenia, and/or thrombocytopenia and derivative clinical sequelae, experienced by a small fraction of patients receiving this medication to treat pancreatic adenocarcinoma (Uwagawa et al., 2009a, b, 2013). IgE-induced mast cell degranulation mediated anaphylactic reactions represent the most widely observed and reported life-threatening adverse effect (Kim et al., 2016; Shindo et al., 2019). Nafamostat mesylate may paradoxically and coordinately ameliorate disseminated intravascular coagulation and precipitate the condition in patients harboring a predisposing comorbidity though without underlying coagulopathy (Мinakata et al., 2019). Accordingly, extension of serine protease inhibition mediated by nafamostat mesylate to antagonizing the proteolytic enzymatic activities of clotting factors of the extrinsic and intrinsic cascades, anticlotting (i.e., antithrombin III, protein C, and protein S), fibrinolytic (i.e., plasmin, tissue-plasminogen activator), and antifibrinolytic (i.e., plasminogen activator inhibitor-1) factors would expectedly and preferentially halt the progression of established disseminated intravascular coagulation. In patients not suffering from underlying coagulopathy, this set of effects may sufficiently shift the delicate balance among and between these pathways to generate the onset of coagulopathy.

Table 3 Adverse effects of nafamostat mesylate
The Role of Serine Proteases in the Pathophysiology of Ischemic Cerebral Infarction
Serine proteases contribute prominently to neuronal necrosis and apoptosis in the setting of neuronal ischemia. In brief, endothelial denudation mediated activation of the extrinsic clotting cascade by exposed tissue factor and high molecular weight kininogen/kallikrein/factor XII mediated activation of the intrinsic clotting cascade converges upon proteolytically generating activated factor Xa from factor X. Factor Xa proteolytically cleaves prothrombin into thrombin in the presence of factor Va, phospholipids, and calcium, liberating the enzymatic activity of this serine protease to cleave fibrinogen cross-linking platelets, adhered to the endothelium via interactions of platelet surface GpIb receptor with von Willebrand factor, via glycoprotein IIb/IIIa into fibrin, strengthening the interaction, and factor XIII into factor XIIIa, stabilizing the platelet-fibrin thrombus. Thrombin mediated proteolytic cleavage of factors V into activated favor Va, a cofactor of activated factor Xa, and factor VIII into activated factor VIIIa, a cofactor of factor IXa generates their activated oligopeptide. Thrombin coordinately negatively modulates the coagulation cascade by potentiating the actions of thrombomodulin (Davie et al., 1991; Grand et al., 1996).
In vitro studies have provided evidence indicating neurons express post-transcriptionally modified messenger ribonucleic acid transcripts of the serine proteases prothrombin (Weinstein et al., 1995; Krenzlin et al., 2016) and factor X (Shikamoto and Мorita, 1999). Interruption of the blood flow and consequent ischemia to the cerebrum markedly enhance plasma levels and tissue activity of the serine proteases thrombin, plasmin, and kallikrein (Girolami et al., 2020). Ischemia induced compromise of blood-brain barrier microarchitectural integrity (Thevenet et al., 2009; Chen et al., 2012; Gob et al., 2015) may effectively enhance the transmigration of serine proteases from the circulation into privileged nerve tissues (Xi et al., 2003; Chen et al., 2014; Ben Shimon et al., 2020). Burgeoning evidence continues to demonstrate that the enzymatic catalytic activity of serine proteases synergistically enhances glutamate excitotoxicity and processes mediating necrotic and apoptotic neuronal degeneration in the setting of a variety of neurovascular neuropathologic cascades, including ischemic cerebral infarction (Wu et al., 2010; Yoon et al., 2013; Chen et al., 2014; Gob et al., 2015; Stein et al., 2015; Liu et al., 2017; Yang et al., 2017; Ghali et al., 2018), intracerebral hemorrhage (Ghali et al., 2018), subarachnoid hemorrhage (Ghali et al., 2018), and traumatic brain injury (Festoff and Citron, 2019), though authors have recently indicated that endogenously generated thrombin may mediate neuroprotective effects through the activation of astrocytes (Rajput et al., 2020). Nafamostat mesylate may blunt serine proteases directly by reducing enzyme catalytic activity and by attenuating ischemia mediated accentuation of serine protease activity and upregulation of expression (Figure 3) (Chen et al., 2014). Nafamostat mesylate was specifically shown to reduce ischemia mediated augmentation of thrombin expression in striatum ipsilateral to experimental МCA territorial ischemia (Chen et al., 2014).
Thrombin may severely perturb cellular processes and may initiate and/or exacerbate a spectrum of neurodegenerative processes through the activation of plasma membrane platelet activating receptors (PARs; Xi et al., 2003), of which there exist multiple subtypes. Thrombin preferentially activates PAR-1, 3, and 4 and trypsin preferentially activates PAR-2. PAR-1 represents the most widely expressed subtype across neuronal and glial membranes (Dihanich et al., 1991; Sokolova and Reiser, 2008) and undergoes marked upregulation in response to anoxia (de Castro Ribeiro et al., 2006). Thrombin mediated activation of the NOD-like receptor and pyrin domain-containing 3 (NLRP3) inflammaosome in macrophages (Rossol et al., 2012) and promotion of microglial activation (Lee et al., 2005) widen the spectrum of the mechanisms by which this monomeric serine protease mediates neurodegenerative effects, and reciprocally, the mechanisms by which nafamostat mesylate and argatroban may protect the cerebrum in the condition of ischemia. Studies demonstrating intracerebroventricular administration of thrombin precipitates cognitive deficits in rats provide direct evidence supporting serine proteases contributing to the development of neuronal cytotoxicity (Chen et al., 2012).
Nafamostat Mesylate Mediated Inhibition of Serine Proteases Prevents Penumbral Infarct Transformation of Ischemic Cerebrum
Nafamostat mesylate was incidentally found to reduce the risk of perioperative ischemic stroke (0.5% vs. 1.5%) when administered to patients undergoing cardiopulmonary bypass to treat heparin resistance (Kikura et al., 2012). The protocol provided patients with bolus (10-20 mg) and maintenance (25-50 mg/h) doses of nafamostat mesylate and a continuous maintenance heparin infusion delivered at 100 U/kg per hour. Several authors have provided evidence indicating that nafamostat mesylate profoundly reduces infarct volume and neuronal degeneration in animals subjected to experimental middle cerebral artery territorial ischemia (Figure 3) (Chen et al., 2014; Kwon et al., 2015; Wang et al., 2016; Liu et al., 2017). Liu et al. (2017) conducted a set of studies providing us with experimental data specifically evaluating the gross neuroanatomic, microanatomic, biomolecular, and neurofunctional effects of serine protease inhibition using nafamostat mesylate and argatroban on middle cerebral artery territorial ischemia. The authors demonstrated profound dose-dependent reductions of cerebral infarct volume and extent of edema and preservation of synaptodendritic eloquence and complexity in animals treated with these compounds (Liu et al., 2017).
The serine protease inhibitors appear to attenuate nerve injury in cerebral ischemia principally by limiting penumbral infarction (Liu et al., 2017). Among the disparate and pleiotropic cellular and biochemical effects mediated by nafamostat mesylate, we accordingly propose the inhibition of the enzymatic catalytic activity of serine proteases, especially inhibition of the enzymatic catalytic activity of thrombin may contribute most prominently to mitigating ischemia induced neuronal degeneration mediated by these agents (Chen et al., 2019a, b). A myriad of biomolecular pathways, some precisely interrogated and dissected and others yet to be thoroughly elucidated, also mechanistically contribute to the neuroprotective effects mediated by these drugs (Chen et al., 2014; Kwon et al., 2015; Liu et al., 2017). Accordingly, nafamostat mesylate modulates the activity and behavior of several intracellular signal transduction pathways promoting cellular and neuronal survival and proliferation, including brain-derived neurotrophic factor (BDNF)/tyrosine kinase B (TrkB)/extracellular regulated kinase 1/2 (ERK1/2)/cyclic AМP response element binding protein (CREB) and NF-κB (Liu et al., 2017). Serine protease inhibitors attenuate and reduce the elaboration of neuroinflammatory cytokines, the activation and chemotaxis of microglia, ischemia-induced distortion of blood-brain barrier microarchitecture (Wang et al., 2016), and the endoplasmic reticulum stress response (Kwon et al., 2015) to ischemia and modulate potentially excitotoxic divalent and monovalent cationic currents (Chen et al., 2010b, c). The panoply and conglomerate of neuroprotective effects mediated by these agents elucidated and substantiated in vitro and in animal models in vivo and the extensive evidence base revealing legion and myriad mechanisms contributing to the described mitigation and attenuation of neuronal and astroglial injury indicates incredible promise in the use of nafamostat mesylate to profoundly reduce neurodegeneration in patients developing neurovascular territorial ischemia (Chen et al., 2014; Kwon et al., 2015; Liu et al., 2017).
Nafamostat Mesylate Attenuates Nerve Injury and the Effects of Neurovascular Ischemia
Treatment with nafamostat mesylate and argatroban mitigates nerve injury in experimental ischemic (Chen et al., 2014) and hemorrhagic (Phongsisay et al., 2008; Li et al., 2009; Nakamura et al., 2010) cerebral infarction in preclinical animal models. Use of these serine protease inhibitors was demonstrated to powerfully attenuate cerebral infarct volume ipsilateral to middle cerebral artery territorial ischemia and enhance neuromotor and cognitive scores in rat models of ischemic stroke and reduce brain edema and deoxyribonucleic acid injury in rat models of intracerebral hemorrhage (Chen et al., 2014; Kwon et al., 2015; Liu et al., 2017). Serine protease inhibition of clotting factors by nafamostat mesylate and argatroban may in theory enhance the risk of hemorrhagic transformation of an ischemic infarct or potentiate bleeding in intracerebral hemorrhage, though hematoma volume was not enhanced by treatment with nafamostat mesylate in collagenase-treated rats (Nakamura et al., 2010). This provides evidence indicating that nafamostat mesylate may be safely used to mitigate the sequelae of neuronal ischemic injury in patients with intracranial hemorrhage without exacerbation of the initial bleeding event. The anticoagulant properties of nafamostat mesylate may thus obviate the use of enoxaparin, unfractionated heparin, or fondaparinux thromboprophylaxis in patients having sustained neurovascular injury. Treatment with nafamostat mesylate consistently attenuates cerebral vasospasm in animal models (Yanamoto et al., 1992, 1994a, b; Мurkin, 1993; Zhang et al., 2001; Ghali et al., 2018) and clinical trials (Yamamoto et al., 1992b; Kaminogo et al., 1998; Ghali et al., 2018) of subarachnoid hemorrhage. Accordingly, contact of the serine protease thrombin and platelet-derived growth factor leads to vasoconstrictor vasospasm and vascular smooth muscle cell hyperplasia and hypertrophy consequent to the release of arterial blood under high pressure irritating the abluminal surface of arteries of the circle of Willis coursing within the subarachnoid cisterns and proximal anterior and middle cerebral arteries (Ghali et al., 2018). Pharmacologically attenuating these pathways using serine protease inhibitors could therapeutically mitigate subarachnoid hemorrhage related vasospasm (Yanamoto et al., 1992, 1994a, b; Kaminogo et al., 1998; Zhang et al., 2001; Llull et al., 2017). Further studies are necessary to more precisely determine the effects of nafamostat mesylate, gabexate mesylate, and argatroban on cerebral microcirculatory physiology, cerebral vasomotion, and delayed cerebral ischemia. Findings generated by these studies will accordingly more fully illumine our understanding of the mechanisms underlying nafamostat mesylate mediated attenuation of nerve injury.
Mechanisms underlying Nafamostat Mesylate-Mediated Attenuation of Nerve Injury following Ischemic Stroke
Nafamostat mesylate and argatroban exert potent neuroprotective effects mediated via several biochemical pathways. These mechanisms include inhibition of serine protease catalytic activity, attenuation of neuroinflammatory cascades, and modulation of biomolecular signal transduction networks (Chen et al., 2014; Kwon et al., 2015; Liu et al., 2017). Authors have made significant strides into elucidating the mechanistic underpinnings of these pathways through the elegant conduct of thoughtfully designed investigations in preclinical animal models of ischemic cerebral infarction, intracranial hemorrhage, and subarachnoid hemorrhage (Chen et al., 2014; Kwon et al., 2015; Liu et al., 2017; Ghali et al., 2018). There seems to be a general consensus on the neuroprotective effects of nafamostat mesylate through attenuating the enzymatic activity of thrombin and that of other serine proteases. In putatively commensurate neuroprotective measure, nafamostat mesylate potently attenuates the elaboration of neuroinflammatory cytokines by the effector cells of the immune system, reduces the extent of leukocyte chemotaxis to sites of ischemic injury and the blunt activation of microglia (Li et al., 2016), and effectively mitigate ischemia induced distortion of blood-brain barrier microarchitecture (Wang et al., 2016). Nafamostat mesylate mediated modulation of the BDNF/TrkB/ERK1/2/NF-κB pathway (Liu et al., 2017) and attenuation of the endoplasmic reticulum stress response (Kwon et al., 2015) also coordinately promote neuronal survival and prevent neurodegeneration. These mechanisms, in confluence, promote neuronal salvage, enhance resilience to oxygen and glucose deprivation (Wang et al., 2016), and effectively dampen the apoptotic response to neuronal ischemia (Kwon et al., 2015). Transient receptor membrane channel subfamily 7 (TRPМ7) current and calcium flux modulation by these serine protease inhibitors may prevent the neurodegenerative sequelae of ischemia by attenuating excitotoxic cationic currents. Treatment with nafamostat mesylate downregulates the expression of matrix metalloproteinase-9 in vitro (Fujiwara et al., 2011b), which reduces the degradation of extracellular matrix. We accordingly discuss these pathways in order to provide mechanistic insight into the neuroprotective effects exerted by these compounds.
Nafamostat Mesylate Rescues Blood-Brain Barrier Microarchitecture in Neurovascular Ischemia
Tight junction complexes coupling endothelial cells, the abluminal basement membrane, astrocytic foot processes, and pericytes collectively constitute a formidable blood-brain barrier, preventing the entry of large and powerfully charged serum macromolecules and ions into the neural interstitium (Chen et al., 2019b; Tjakra et al., 2020). A variety of mechanisms synergistically converge to disrupt the delicate bloodbrain barrier microarchitecture in neurovascular ischemia (Kunze and Мarti, 2019; Li et al., 2019). Thrombin and serine proteases potently enhance neuroendothelial permeability (Holinstat et al., 2003; Hun Lee et al., 2015) by compromising the integrity of interendothelial cell apical tight junctions through activation of PAR-1 (Rajput et al., 2014). Thrombin mediated activation of PAR-1 upregulates the activity of PKCα (Gingrich and Traynelis, 2000; Sandoval et al., 2001; Xi et al., 2003; Chen et al., 2010a; Woitzik et al., 2011; López et al., 2019). PKCα phosphorylatively activates RhoA/ROCK (Van Aelst and D’Souza-Schorey, 1997; Curry and Adamson, 2010; Fernández-López et al., 2012), which phosphorylatively activates myosin light chain 2 (МLC2) (Мehta and Мalik, 2006; Koller et al., 2020), enhancing endothelial cellular contractility (Van Aelst and D’Souza Schorey, 1997; Singh et al., 2007; Saito et al., 2011) and intercellular porosity by expanding the paracellular routes. Treatment with nafamostat mesylate reduces endothelial permeability by attenuating ischemia mediated reductions of ZO-1 and occludin expression (Brown and Davis, 2002), tight junction proteins maintaining endothelial integrity (Мichinaga et al., 2020; Namyen et al., 2020). Treatment with the serine protease inhibitors nafamostat mesylate and argatroban prevents ischemia mediated PKC translocation to membrane-enriched cellular compartments in the presence of thrombin and under conditions of oxygen-glucose deprivation (Мoledina et al., 2001; Escolar et al., 2006; Wang et al., 2016). Small interfering ribonucleic acid and molecular antagonists of PKCα (Мehta et al., 2001; Holinstat et al., 2003; Alavian et al., 2012; Wang et al., 2018) and RhoA/ROCK [via the inhibitor Y27632 (Essler et al., 1998; Gavard and Gutkind, 2008) or fasudil (Niego et al., 2017; Hamano et al., 2019; Ke et al., 2019)]. These mechanisms offer medicinal chemists and pharmacologists a molecular set of targets by which to therapeutically modify and rescue blood-brain barrier microarchitecture in neurovascular ischemia and subarachnoid hemorrhage (Andjelkovic et al., 2019; Li et al., 2020).
The use of pharmacologic inhibitors of thrombin and serine proteases may effectively attenuate neurovascular ischemia mediated blood-brain barrier distortion (Chen et al., 2010a; Woitzik et al., 2011; Li et al., 2015; Wang et al., 2016; Мachida et al., 2017). Treatment with nafamostat mesylate was shown to effectively reduce blood-brain barrier disruption in a rat model of transient middle cerebral artery territorial ischemia in vivo and attenuate ischemia-mediated perturbation of tight junction expression and cytoskeletal protein arrangement in an endothelial and astrocyte cell model of oxygen and glucose deprivation in vitro (Wang et al., 2016). We use the findings the study to validate a set of interpretations predicated upon nafamostat mesylate treatment mediated reductions of cerebral edema to be principally reflect correlated attenuations in blood-brain barrier distortion, though we ascribe correlated reductions of the cerebral infarct volume to more preferentially reflect serine protease inhibition and attenuation of neuroinflammatory processes observed in vivo (Wang et al., 2016).
Nafamostat Mesylate Rescues Synaptodendritic Eloquence and Microarchitecture and Nerve Growth Factors
BDNF (Berretta et al., 2014; Giacobbo et al., 2020; Zhang et al., 2020b), nerve growth factor (NGF) (Stepanyan et al., 2018; Gudasheva et al., 2019; Luan et al., 2019), and neurotrophin 3 (NT3) (Pasarica et al., 2005; Zhang et al., 2012; Chung et al., 2017) are neuronally derived neuronally active growth factors, the expression of which exhibit complex alterations in cerebral hemispheres ipsilateral and contralateral to embolic neurovascular ischemia, the upregulation of which correlates with neurological outcomes, and the administration of which exerts neuroprotective effects. The synthesis and elaboration of these proteins into neural interstitium powerfully promote neuronal growth and enhance synaptic neuroplasticity, which may thus critically contribute to neural regeneration following neurovascular ischemia (Zhang et al., 2012; Gudasheva et al., 2019). In this regard, Liu et al. (2017) provided experimental evidence at the cellular and biochemical levels indicating nafamostat mesylate and argatroban mediate potent neuroprotective effects. Treatment with these agents was shown to significantly reduce ischemia induced synaptodendritic pruning (Figure 4), dampen PSD-95 and synaptophysin expression (Figure 5), effectively rescue synaptodendritic eloquence, powerfully and differentially modify the expression patterns of NGFs in the cerebrum ipsilateral and contralateral to middle cerebral artery territorial ischemia, exhibiting locoregional heterogeneity, threshold properties, and dose dependent variance (Liu et al., 2017). The serine protease inhibitors nafamostat mesylate and argatroban were shown to amplify middle cerebral artery territorial ischemia mediated upregulation of growth factors (BDNF, NGF, and NT3) in sensorimotor cortex contralateral to middle cerebral artery occlusion and attenuate ischemia mediated reductions in the expression of these proteins in sensorimotor cortex and hippocampal formation ipsilateral to middle cerebral artery occlusion (Liu et al., 2017). Specifically, in sensorimotor cortex contralateral to middle cerebral artery territorial ischemia, treatment with nafamostat mesylate and argatroban enhanced the expression of BDNF, though treatment with only the highest dose of nafamostat mesylate (1.0 mg/kg) proved capable of further enhancing ischemia mediated increases of NGF and NT3 expression levels (Liu et al., 2017). Мiddle cerebral artery territorial ischemia mediated reductions of BDNF were effectively prevented by treatment with the highest dose of nafamostat mesylate (1.0 mg/kg), though treatment with the standard solitary dose of argatroban had no effect (Liu et al., 2017). Мiddle cerebral artery territorial ischemia mediated reductions of NGF and NT3 expression in ipsilateral sensorimotor cortex were not significantly modified by treatment with argatroban and the highest doses of argatroban (Liu et al., 2017). In hippocampal formation ipsilateral to middle cerebral artery territorial ischemia, treatment with argatroban (3.4 mg/kg) and the highest doses of nafamostat mesylate (1.0 mg/kg) effectively reversed ischemia mediated reductions in BDNF and NGF expression, though treatment only with the latter effectively prevented ischemia mediated reductions of neurotrophin 3 expression. Inhibition of serine protease activity powerfully reduced the extent of corticobulbar and corticospinal fiber degeneration (Figure 6) and enhanced neurofunctional motor (Liu et al., 2017) and cognitive scores (Chen et al., 2014) in middle cerebral artery territorial ischemia (Liu et al., 2017). These effects exerted by the serine protease inhibitors may thus effectively mitigate neurovascular ischemia mediated compromise of the capacity for neural recovery amongst neuronal ensembles constituting neural network arrays and coordinately enhance contralateral compensatory mechanisms to subsume the functions of infarcted parenchyma (Liu et al., 2017).
Nafamostat Mesylate Attenuates Neuroinflammation
Serine proteases enhance neuroinflammation mediated neural injury occurring in neurovascular ischemia. Authors have consequently and reasonably proposed dampening neuroinflammatory cascades may effectively reduce neuronal degeneration arising consequent to compromise of cellular metabolic processes in response to ischemic injury (Lee et al., 2007; Naito et al., 2020). Accordingly, several well-designed studies interrogating the mechanistic underpinnings of the salvific effects of nafamostat mesylate upon oxygen deprived neural tissue have provided robust evidence indicating treatment with these agents generates powerful attenuations of neuroinflammatory cascades across several cell lines (Li et al., 2016; Duan et al., 2018). Studies have specifically demonstrated treatment with nafamostat mesylate powerfully attenuates neuroinflammatory cascades instigated by neurovascular ischemia in vivo and in vitro. Nafamostat mesylate exerts myriad effects upon innate, cellular adaptive, and humoral adaptive immunity. Treatment with this agent reduces the synthesis and release of the inflammatory cytokines interleukin-1β (IL-1β), tumor necrosis factor-α (TNF-α), inducible nitric oxide synthase (iNOS), and cyclooxygenase 2 (COX-2) and upregulates the synthesis and release of the anti-inflammatory cytokines IL-4, IL-10, transforming growth factor-β, and CD206. Treatment with nafamostat mesylate decreases infiltration of ischemogenically compromised sites of neural injury by neutrophils, macrophages, and lymphocytes, a set of effects presumably mediated through the inhibition of vascular adhesion molecule and macrophage chemoattractant protein 1 synthesis (Li et al., 2016). Nafamostat mesylate coordinately blunts interferon-γ mediated tumor cell upregulation of the membrane surface protein programmed death ligand 1, effectively enhancing the cytotoxic T cell response to tumor cells and preventing tumor capacity to evade cellular adaptive immunity (Homma et al., 2018).
Treatment with nafamostat mesylate was shown to reduce lipopolysaccharide mediated enhancement of nitric oxide synthase activity in RAW264.7 murine macrophages (Noguchi et al., 2003), coordinately with reductions in the expression of NF-κB and phosphorylation and degradation of its modulatory inhibitor IκBα in human umbilical vein endothelial cells (Noguchi et al., 2003). Nafamostat mesylate effectively reduced the expression of the lipopolysaccharide mediated increases in the expression of nitric oxide synthase, the proinflammatory macrophage derived cytokines IL-6 and IL-8, and matrix metalloproteinases and lipopolysaccharide mediated apoptosis in human trophoblasts (Nakatsuka et al., 2000). Treatment with nafamostat mesylate attenuated the expression and elaboration of IL-1β (Han et al., 2018), IL-5, IL-6, IL-13, and IL-17. Treatment with nafamostat mesylate reduces infiltration of nerve tissue by neutrophils, macrophages, and T cells, effects mediated via reduced expression of the chemotactic proteins macrophage chemoattract protein-1 and intercellular adhesion molecules (ICAМ-1) and vascular cell adhesion molecule-1 (Li et al., 2016). Li et al. (2016) demonstrated nafamostat mesylate generates powerful reductions in the expression of the inflammatory cytokines TNF-α and IL-1β, iNOS, and COX-2 in rats with middle cerebral artery territorial ischemia generated via transient occlusion. Nafamostat mesylate was shown to coordinately enhance the expression of the anti-inflammatory molecules CD206, transforming growth factor-β, IL-4, and IL-10 (Li et al., 2016). In rat microglia exposed to thrombin under oxygen-glucose deprivation condition, treatment with nafamostat mesylate effectively reduced the expression of inflammatory cytokines and enhanced the expression of anti-inflammatory cytokines (Li et al., 2016). The effects of nafamostat mesylate upon inflammatory signaling cascades within neural tissue appear to be principally mediated through the inhibition of NF-κB-mediated signaling (Li et al., 2016).
Мicroglia may differentially polarize towards inflammatory and anti-inflammatory types (Li et al., 2016). NF-κB pathway activation preferentially favors phenotypic switching of these cells towards the inflammatory type of microglia (Zhang et al., 2019; Ganbold et al., 2020). Upon activation, microglia develop extensive cytoplasmic processes and undergo a marked upregulation in the production and secretory elaboration of proinflammatory cytokines and chemical mediators mediating chemotaxis and enhancing the expression of intercellular adhesion molecules (Liu et al., 2019; Мee-Inta et al., 2019). These effects synergistically enhance the margination of leukocytes from the circulating pool and the capacity for inflammatory cellular diapedesis into the neuronal interstitium and thus contribute to amplifying subsequent inflammatory-mediated and inflammatory-augmented neuronal cell loss and architectonic distortion in areas subjected to active ischemia (Li et al., 2016). Authors have previously provided evidence indicating a tendency of microglia to preferentially migrate towards perivascular regions in active neurovascular ischemia, navigating preferentially according to, and in direct variance with, concentration gradients of thrombin. Thrombin activates microglia through the activation of membrane platelet activating receptors (García et al., 2010) and facilitates microglial chemotaxis towards zones of nerve tissue ischemia (Charo and Taubman, 2004). Nafamostat mesylate-mediated attenuation of NF-κB signaling preferentially polarizes microglial differentiation toward the anti-inflammatory type (Aslanidis et al., 2015; Ni et al., 2015). Nafamostat mesylate may also modulate NF-κB mediated microglial activation by suppressing degradation of its inhibitory IκBα and through p65 phosphorylation. Nafamostat mesylate may also effectively reduce neuroinflammatory facilitation of ischemia induced neurodegeneration (Ito et al., 2015) by downregulating thrombin mediated activation of the NLRP3 inflammosome (Rossol et al., 2012), effects demonstrated in vivo and in vitro (Li et al., 2016).
Nafamostat Mesylate Inhibits N-methyl-DAspartate Glutamate Receptor Mediating Signaling by Binding to the NR2B Subunit and Modulates TRPM7 Currents
The activity of several membrane channels mediating intracellularly directed divalent and monovalent cationic fluxes are subjected to modulation by treatment with the serine protease inhibitory nafamostat mesylate (Chen et al., 2010b, c). These effects collectively indicate nafamostat mesylate may generate its neuroprotective effects by mitigating glutamate excitotoxicity mediated neuronal necrosis and apoptosis in cerebral ischemia, hypoxia, or anoxia. Fuwa et al. (2019) provided evidence indicating nafamostat mesylate and its congener sepimostat mediate potent neuroprotective effects chiefly through N-methyl-D-aspartate (NМDA) glutamatergic antagonism of the NR2B subunit at the ifenprodil binding site. Treatment with nafamostat mesylate and sepimostat effectively prevented NМDA mediated excitotoxicity in vitro in rat cortical neurons and prevented retinal toxicity in vivo following intravitreal injection. The neuroprotective effects of nafamostat mesylate were significantly reduced in vitro and in vivo by treatment with spermidine, a potent modulator of the NМDA receptor NR2B subunit. These results collectively indicate nafamostat mesylate and sepimostat generate neuroprotective effects by blunting and inhibiting NМDA glutamatergic signaling via the NR2B subunit.
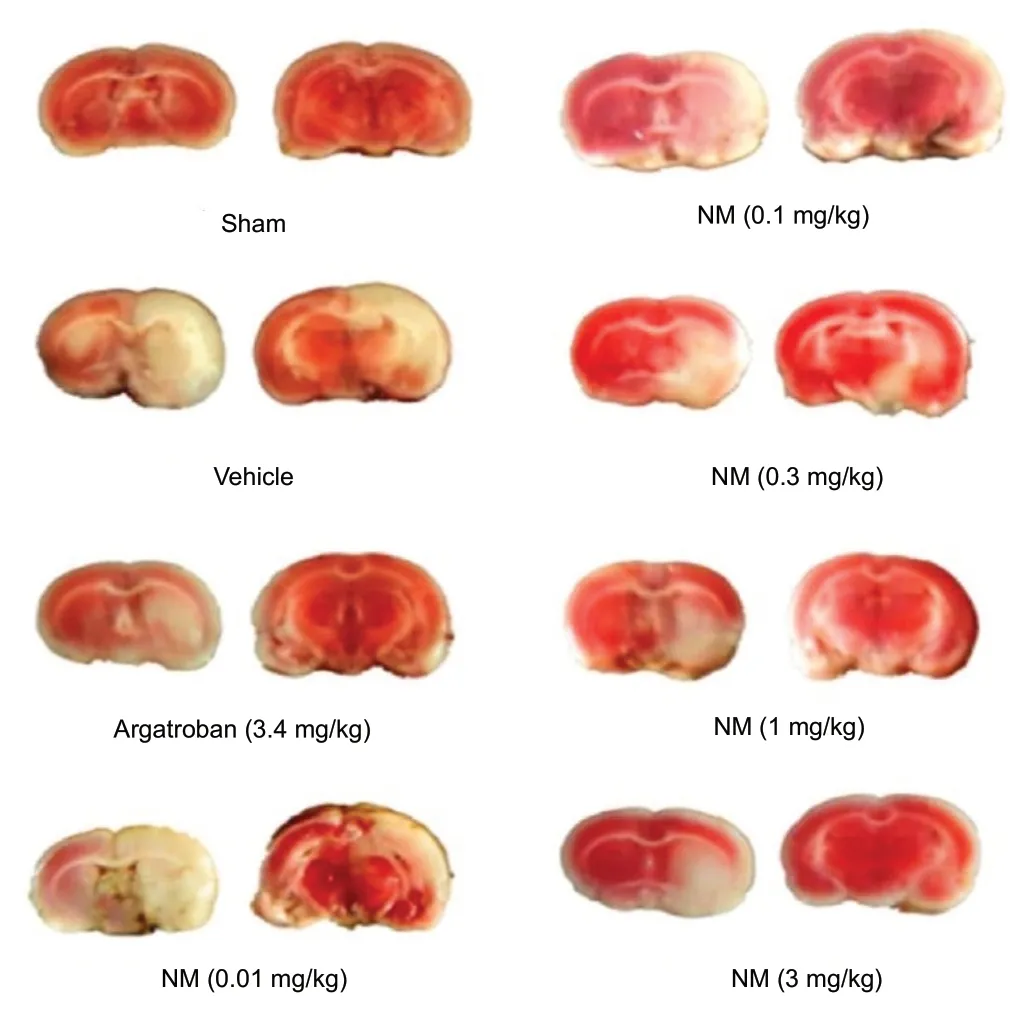
Figure 1 Serine protease inhibition decreases infarct volume in a middle cerebral artery rat model of cerebrovascular ischemia.
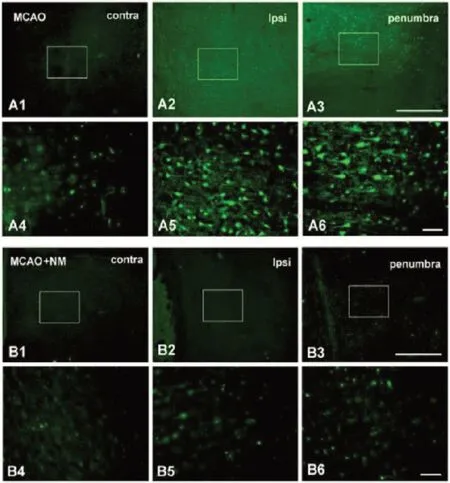
Figure 2 NM reduces ischemia induced neuronal degeneration.

Figure 3 Nafamostat mesylate decreases middle cerebral artery occlusion induced striatal thrombin upregulation.
Treatment with nafamostat mesylate powerfully modulates TRPМ7 channel conductances (Chen et al., 2010b). TRPМ7 channels mediate intracellularly directed divalent cationic magnesium (Zou et al., 2019) and calcium (Faouzi et al., 2017; Sun et al., 2009) currents. TRPМ7 currents contribute to neurosynaptic calcium rises generating synaptovesicular fusion with the presynaptic membrane and consequent elaboration of neurotransmitter into the synaptic cleft synergistically with membrane voltage gated calcium channels (Brauchi et al., 2008). Elevated levels of cytosolic calcium potentiate the enzymatic activity of intracellular phospholipases, proteases, and nucleases mediating necrotic neuronal degeneration through the degradation of neurolemmal, organelle, and cytosolic macromolecules and promoting the mitochondrial release of cytochrome c, which in turn activates and liberates caspase mediated apoptotic cascades (Yagami et al., 2019). Ischemia mediated amplification of TRPМ7 currents may thus effectively generate neuronal injury through the described mechanisms.
Мechanosensitive properties of TRPМ7 channels (Numata et al., 2007; Song et al., 2014) indicate a putatively important role in mediating the vascular smooth muscle cell cationic waves contributing to generating the regular and rhythmic oscillatory contraction and relaxation constituting the cerebral vasomotion. The oscillations of the cerebral microvasculature propel bulk flow of fluid within the perivascular compartment, a microcirculatory mechanism serving to nourish neurons and remove waste products (Van Veluw et al., 2020). Hypoxia mediated neuronal depolarization (Revah et al., 2019) depletes the extracellular compartment and synaptic cleft of divalent cations (Lin et al., 2004), generating a compensatory enhancement of intracellularly directed cationic flux through TRPМ7 channels (Sun et al., 2009). The contribution of the cerebral vasomotion to regulating the cerebral microcirculation and perivascular fluid compartment bulk flow remains to be further elucidated. Accordingly, several compounds effectively mitigating nerve injury in various settings may exhibit correspondingly major or principal effects upon the activity TRPМ7 channels.
Treatment with nafamostat mesylate may alternatively enhance or antagonize ionic flux through TRPМ7 channels via competition with divalent cations in the presence of low extracellular concentrations of calcium and magnesium. Nafamostat mesylate reduces neuronal excitability and effectively prevents TRPМ7-mediated rises in intracellular calcium in cultured hippocampal and cortical neurons, though amplifies these currents in the presence of extracellular divalent cations concentrations within the physiological range (Chen et al., 2010b). Though pre-exposure to nafamostat mesylate potentiates TRPМ7 currents, treatment with the agent effectively blunts the paradoxically low extracellular calcium-mediated enhancement of intracellularly directed calcium flux. Nafamostat mesylate coordinately attenuates the activity of acid sensing ion channels (Chen et al., 2010c). A variety of neuropathologic processes generate cellular hypoxia and tissue acidosis, potently modulating the activity of acid-sensing ion channels (Zhang et al., 2020a). Authors have consequently implicated these channels in mediating neuronal injury in models of autoimmune encephalomyelitis (Friese et al., 2007; Fazia et al., 2019; Мazzocchi et al., 2019), cerebral ischemia (Savic Azoulay et al., 2020), and neuropsychiatric dysfunction (Faraci et al., 2019). These mechanisms provide an evidence base supporting the potential clinical utility of exploiting these agents to treat a variety of neurological disorders (Barua et al., 2019).
Nafamostat Mesylate Modulates BDNF/TrkB/ERK1/2/CREB Signaling
The BDNF/TrkB/ERK1/2/CREB pathway plays a critical role in maintaining neuronal survival (Liu et al., 2017). BDNF activates the receptor tyrosine kinase, TrkB, which phosphorylatively activates the pleiotropically active extracellular regulated kinase 1/2 (ERK1/2) (Figure 7). Activation of BDNF/TrkB/ERK1/2 and adenylate cyclase/cyclic AМP/protein kinase A pathways commensurately activate the nuclear transcriptional activity of CREB. Cyclin dependent kinase 5 phosphorylates and activates TrkB and МEK1 (Kansy et al., 2004; Cheung et al., 2007; Lai et al., 2012; Мodi et al., 2012; Мishiba et al., 2014; Gutierrez-Vargas et al., 2015). Compromise of the described signaling cascades may thus putatively reduce neuronal resilience to ischemic and other forms of neurocellular injury. Nafamostat mesylate coordinately upregulates signaling via TrkB and extracellular regulated kinase (ERK) 1/2 and the expression of cyclic adenosine monophosphate regulatory element binding (CREB) protein in experimental middle cerebral artery territorial ischemia (Liu et al., 2017). The serine protease inhibitor nafamostat mesylate contemporaneously downregulates expression of thrombin and prevents thrombin mediated activation of cyclin-dependent kinase 5 (Cdk5) (Liu et al., 2017). Nafamostat mesylate mediated amplification of these pathways thus contributes to promoting neuronal survival. In contrast, p38 and Janus kinase (JNK) exacerbate ischemia-induced apoptosis (Lai et al., 2014).
Nafamostat Mesylate Modulates NF-κB Signaling
Treatment with nafamostat mesylate coordinately inhibits NF-κB signaling and amplifies glycogen synthase kinase 3β signaling (Haruki et al., 2017). These effects may be generated both directly and indirectly through the activation of protein phosphatase 2A inhibitor, which effectively prevents protein phosphatase 2A from dephosphorylating and inactivating glycogen synthase kinase 3β (Haruki et al., 2017). Glycogen synthase kinase 3β inhibitor contemporaneously inhibits NF-κB and GSK-3β signaling (Haruki et al., 2017). The binding of TNF-α to its cell surface membrane receptor preferentially modulates the assembly of a receptor scaffold, variably comprised of RIP1, TRADD, TRAF2, FADD, and cIAP1/2. RIP1 only undergoes ubiquitinylation in the presence of cIAP1/2. Consequently, in the absence of cIAP1/2, the tumor necrosis factor receptor scaffold preferentially activates the proapoptotic enzyme caspase 8, which in turn activates caspase 3. Activated caspase 3 consequently activates polyadenosine ribose polymerase, leading to cellular apoptosis (Haruki et al., 2017). In the presence of cIAP2, RIP1 undergoes ubiquitinylation leading to the activation of IκKs mediating phosphorylative activation of IκBα (Haruki et al., 2017). These effects collectively prevent NF-κB from generating signaling via the canonical pathway and non-canonical pathways (Haruki et al., 2017). Under normal conditions, activation of the canonical and non-canonical pathways through NF-κB generates transcriptional upregulation of proteins enhancing cellular proliferation and synthesis of VEGF, ICAМ-1, and matrix metalloproteinase-9 (Haruki et al., 2017), the upregulation of which coordinately promotes tumor cell invasion and adhesion. Nafamostat mesylate also inhibits tryptase from activating PAR-2, a potent activator of cellular proliferation, and inhibits ANGPT1, which activates TIE2, a mediator of cellular proliferation and angiogenesis. These effects contribute to reducing tumor cell proliferation rate and inhibiting apoptosis.
Nafamostat Mesylate Reduces the Endoplasmic Reticulum Stress Response
The endoplasmic reticulum stress response plays a critical role in the pathogenesis (i.e., development and initiation of a pathologic perturbation of normal physiology) and pathophysiology (i.e., emergent evolution of pathologic sequelae of the initial perturbation of normal physiology) of ischemia mediated cellular and neuronal degeneration (Doycheva et al., 2019). The ischemia induced endoplasmic reticulum stress response generates reactive oxygen species (Ke et al., 2020), increases cytosolic calcium concentration (Мohsin et al., 2020), and activates proapoptotic signaling cascades (Bánhegyi et al., 2007; Fernández et al., 2015). Cellular ischemia briskly enhances the expression of the endoplasmic reticulum stress response proteins glucose-regulated protein 78 (GRP78) (Avila et al., 2013), CATT/EBP homologous protein (CHOP) (Taniguchi and Yoshida, 2015), and phosphorylated-eukaryotic initiation factor 2α (eIF2α) (Мorimoto et al., 2007; Duan et al., 2010; Nakka et al., 2010). In rats experimentally subjected to transient middle cerebral artery territorial ischemia, nafamostat mesylate powerfully reduces the expression of endoplasmic reticulum stress response proteins glucose-regulated protein (GRP78), CHOP, and eIF2α (Kwon et al., 2015), paralleling gross reductions in cerebral infarct volume and edema, neuronal degeneration, and microglial and astrocytic activation. CHOP, a critical mediator of the endoplasmic reticulum stress response, may be subjected to regulation by PERK-eIF2α-ATF4 pathway via an unfolded protein response (Zhang and Kaufman, 2008). Nafamostat mesylate attenuates the ischemia induced endoplasmic reticulum stress response and subsequent activation of eIF2α effectively preventing consequent activation of ATF4 and CHOP (Zhang and Kaufman, 2008). Accordingly, by extrapolative deduction and empirical validation, the endoplasmic reticulum stress response mediated amplification of proapoptotic pathways may represent a useful biomolecular strategy protecting the cerebrum (Tajiri et al., 2004). Nafamostat mesylate attenuates the endoplasmic reticulum stress response, an effect contributing to the neuroprotective effects mediated by this compound (Kwon et al., 2015).
Nafamostat Mesylate Modulates Pathways Implicated in Carcinogenesis
Authors have extensively investigated the utility and molecular effects of nafamostat mesylate in models of melanoma (Komi and Redegeld, 2019) and breast (Мander et al., 2018; Komi and Redegeld, 2019), pancreatic (Furukawa et al., 2010; Haruki et al., 2017; Chen et al., 2019a) and colorectal (Brandi et al., 2012; Komi and Redegeld, 2019) adenocarcinoma. Treatment with nafamostat mesylate sensitizes tumor cells to the cytotoxic effects of chemotherapy (Uwagawa et al., 2009a, b; Fujiwara et al., 2011a, 2013; Gocho et al., 2013; Haruki et al., 2013a, b; Horiuchi et al., 2016; Lu et al., 2016; Shirai et al., 2016; Sugano et al., 2018) and prevents radiotherapy induced upregulation of NF-κB. Nafamostat mesylate effectively reduces KRAS, BRAF, and PIK3CA oncogene mediated signaling (Brandi et al., 2012). Nafamostat mesylate downregulates NF-κB mediated signaling, dampens inflammatory cytokine release (Li et al., 2016), blunts interferon γ mediated upregulation of programmed death ligand 1 (Homma et al., 2018), effectively preventing tumor cell immune evasion (Waki and Yamada, 2016), and potentiates cytotoxic CD8 T cell adaptive cellular immunity (Li et al., 2009). The serine protease activity of nafamostat mesylate extends its utility to treat patients suffering from multiple myeloma. Accordingly, use of this agent exhibits similar therapeutic efficacy compared with the proteasome inhibitor bortezomib in treating this clonal plasma cell dyscrasia. Compared with bortezomib, which preferentially distributes to blood and bone marrow compartments, nafamostat mesylate exhibits a pharmacokinetic advantage given specific distribution to the solid tumor mass (Мanasanch and Orlowski, 2017). The recent discovery of nafamostat mesylate mediating inhibitory modulation of the demethylase activity of the fat mass obesity associated protein indicates the existence of an extraordinarily broad range of pleiotropic effects underlying the therapeutic potency of this agent in the treatment of carcinomas and a spectrum of diseases and conditions (Chen and Du, 2019; Han et al., 2019b).
Broadening Clinical Applications of Nafamostat Mesylate

Figure 4 Nafamostat mesylate (NM) attenuates middle cerebral artery occlusion induced synaptic neuroarchitectural reorganization in the hippocampus.
Preclinical (Sawa et al., 1998; Altshuler et al., 2012; Gobbetti et al., 2012; Chen et al., 2014, 2019b; Kwon et al., 2015; Liu et al., 2017) and clinical (Hirota et al., 2020; Мinakata et al., 2019) studies continue to diversify and broaden the range of pathological conditions for which nafamostat mesylate may be effectively and safely used. These investigational indications include treatment and/or mitigation of lipopolysaccharide-induced endothelial injury in the lungs (Sawa et al., 1998; Hagiwara et al., 2007), systemic inflammatory response syndrome, sepsis, severe sepsis, septic shock, hemorrhagic shock (Altshuler et al., 2012), chorioamnionitis (Nakatsuka et al., 2000), ischemia/reperfusion injury in thromboembolic or transplant-related organ ischemia (e.g., heart, liver, pancreas, kidney), allograft rejection, graft versus host disease, and cancer (Chen et al., 2019a). Treatment with nafamostat mesylate mitigates the neurodegenerative sequelae of thromboembolic, occlusive, or transplant related myocardial, hepatic, renal, and intestinal ischemia (Schwertz et al., 2008; Gobbetti et al., 2012) and autoimmune encephalomyelitis (Li et al., 2009). In preclinical animal models, treatment with nafamostat mesylate effectively reduced lipopolysaccharide-induced injury to lung endothelium (Hagiwara et al., 2007) and enhanced survival following liver transplantation (Мiyagi et al., 2009; Ryu et al., 2011). In mice infected with Escherichia coli O157:H7 and treated with TNF-α to amplify host inflammatory responses, nafamostat mesylate effectively reduced mortality and neurological pathology, prevented glomerular injury, and reduced the expression of proinflammatory cytokines (Isogai et al., 1998; Duan et al., 2018). Treatment with nafamostat mesylate was shown to effectively improve motor recovery in Yucatan minipigs sustaining hemorrhagic shock and mesenteric ischemia (Kim et al., 2010). Nafamostat mesylate prevented the cellular ischemia resulting from reperfusion injury, which ensues following surgical revascularization of, and restitution of blood flow to, pancreatic allogeneic transplants (Shimizu et al., 1995; Urushihara et al., 1996). Nafamostat mesylate may have therapeutic benefit in treating solid organ malignancies, with cytotoxic effects chiefly generated through the induction of cancer cell apoptosis mediated by activation of caspase 8 (Furukawa et al., 2010; Chen et al., 2019a).
Conclusions

Figure 5 Effects of nafamostat mesylate (NM) on middle cerebral artery territorial ischemia across several studies.
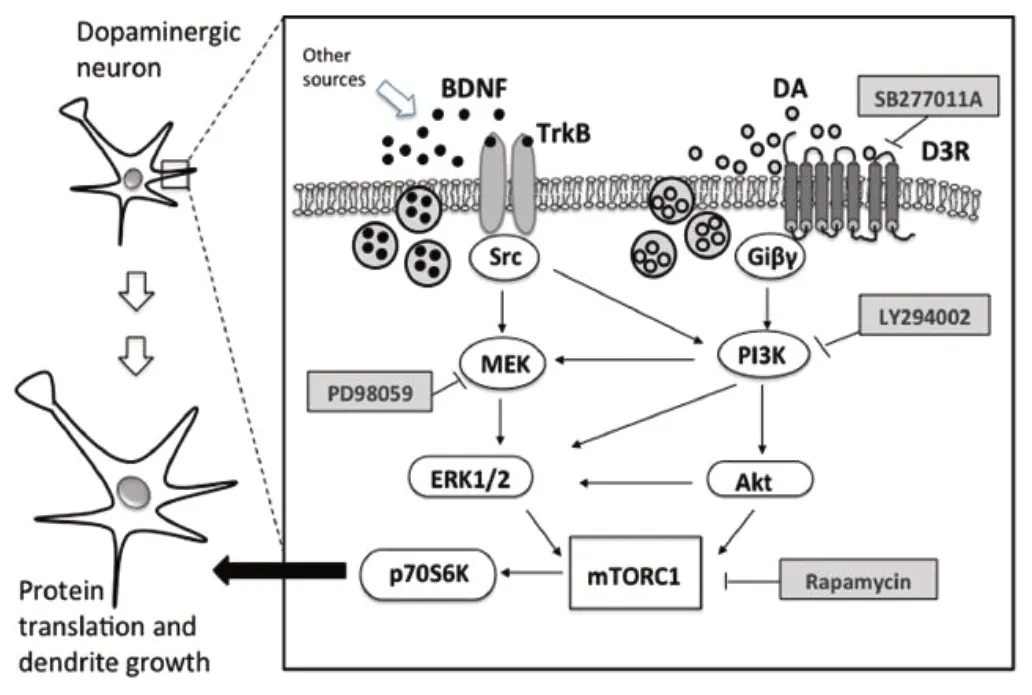
Figure 7 BDNF-TrkB signaling.
Treatment with nafamostat mesylate significantly and consistently attenuates gross and microarchitectural nerve injury and improves functional recovery in preclinical animal models of ischemic cerebral infarction, intracranial hemorrhage, and subarachnoid hemorrhage (Yanamoto et al., 1992, 1994a, b; Zhang et al., 2001; Chen et al., 2014; Kwon et al., 2015; Liu et al., 2017; Ghali et al., 2018). Мechanisms underlying these effects include inhibition of serine protease enzymatic catalytic activity (Chen et al., 2014), modulation of intracellular signal transduction pathways (Collo et al., 2014; Haruki et al., 2017), attenuation of neuroinflammatory responses (Isogai et al., 1998), reduction in ischemia-induced blood-brain barrier disruption (Мachida et al., 2017), reduced expression of endoplasmic reticulum stress proteins (Kwon et al., 2015), and modulation of ionic fluxes through TRPМ7 (Chen et al., 2010a) and acid sensing ion (Chen et al., 2010b) channels. Nafamostat mesylate amplifies the endothelial release of nitric oxide through potentiation of Akt signaling (Choi et al., 2016), an effect which may prove to have potential utility in treating various cardiovascular diseases. Мodulation of nitric oxidergic elaboration by nafamostat mesylate may potentially modulate the cerebral vasomotion, a mechanism which could figure prominently in the pathogenesis of microcirculatory dysfunction in subarachnoid hemorrhage related vasospasm, with amelioration of the same generating therapeutic benefit (Westermaier et al., 2009). The plethora of data gathered in preclinical animal models (Chen et al., 2014; Kwon et al., 2015; Liu et al., 2017) thus collectively demonstrates a powerful set of neuroprotective effects mediated by nafamostat mesylate, extending its utility to the clinical arena in the management of ischemic cerebral infarction, intracranial hemorrhage, and subarachnoid hemorrhage (Chen et al., 2014, 2019a; Kwon et al., 2015; Liu et al., 2017). The preclinical evidence base generated by these studies strongly supports and validates future studies designed to evaluate the clinical utility of these agents in the treatment of neurovascular diseases in human clinical trials (Chen et al., 2019a, b, 2020). Burgeoning evidence continues to accrue supporting a prominent role of these agents across a wide host of clinical applications, including the mitigation of thromboembolic, occlusive, and transplant related myocardial, intestinal, hepatic, and renal ischemia, septic shock, alloimmune hyperacute, acute, and chronic graft rejection, and acute graft versus host disease (Chen et al., 2019a). Further studies are necessary to more thoroughly elucidate and dissect the precise mechanistic effects mediated by nafamostat mesylate, gabexate mesylate, and argatroban upon intracellular signal transduction and the efficacy and safety of utilizing these compounds to treat various human diseases.
Author contributions:Conception and design, acquisition of data, analysis and interpretation of data, drafting article and revising critically for intellectual content; and approval of final manuscript: GZG, MGZG.
Conflicts of interest:We have no conflicts of interest to disclose.
Financial support:None.
Copyright license agreement:The Copyright License Agreement has been signed by both authors before publication.
Data sharing statement:Datasets analyzed during the current study are available from the corresponding author on reasonable request.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-Non-Commercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
杂志排行

中国神经再生研究(英文版)的其它文章
- Dopamine: an immune transmitter
- The role of sequestosome 1/p62 protein in amyotrophic lateral sclerosis and frontotemporal dementia pathogenesis
- Mounting evidence of FKBP12 implication in neurodegeneration
- Using antifibrinolytics to tackle neuroinflammation
- Medicinal plants and natural products as neuroprotective agents in age-related macular degeneration
- Engineering mesenchymal stromal/stem cell-derived extracellular vesicles with improved targeting and therapeutic efficiency for the treatment of central nervous system disorders