采用优化的数字PCR方法分析转基因小麦外源基因拷贝数
2020-06-03琚鹏举宁蕾葛林豪许成杰史华伟梁凯歌马亮刘陶然陈明孙黛珍
琚鹏举,宁蕾,葛林豪,许成杰,史华伟,梁凯歌,马亮,刘陶然,陈明,孙黛珍
(1山西农业大学农学院,山西太谷 030800;2中国农业科学院作物科学研究所,北京 100081;3中央民族大学,北京 100081;4石家庄市农林科学研究院,石家庄 050047)
0 引言
【研究意义】目的基因的拷贝数一直是植物基因组研究的重要内容。外源基因整合进入受体基因组的位置和拷贝数会影响目的基因的表达以及遗传稳定性,而插入基因的拷贝数相对其插入位点而言影响更为重要[1]。当外源基因以低拷贝数(1—2个)整合到受体基因组时,通常能够稳定高效转录表达,多拷贝数的整合则会造成基因不稳定表达,甚至沉默[2]。因此,鉴定转基因产品的外源基因插入拷贝数非常重要[3]。由于小麦基因组庞大复杂,优化和建立高通量拷贝数分析方法对于对小麦转基因育种研究具有重要意义。【前人研究进展】近年来,微滴式数字 PCR(droplet-based digital PCR,ddPCR)是最新兴起的一项核酸检测技术[4-6]。该技术不依赖任何校准物,采用直接计数单个分子的原理,实现了对核酸检测的绝对定量[7-8]。ddPCR已被广泛应用于生命科学研究的多个领域,如核酸绝对定量、稀有突变检测和拷贝数检测等[9]。ddPCR具有高精确度的特点,在检测目的基因拷贝数时,利用目标基因与参考基因的双重反应,通过计算它们的比值,即可快速获得目标基因拷贝数。这种方法由于试验结果重复性好,已经成为了检测基因拷贝数的首选方法[10]。与传统方法相比较,ddPCR具有明显优势,主要体现在操作流程更加简单,无需构建标准曲线,且是对待检测的核酸分子直接进行绝对定量,试验结果更加准确可靠[11]。此外,ddPCR在线性范围、检测极限和定量极限等方面均优于qRT-PCR方法[12]。诸多研究表明,ddPCR已用于玉米、水稻等作物转基因插入拷贝数的检测[13-14]。SUN等[15]的研究表明运用 ddPCR技术可以在甘蔗复杂基因组中明确确定内源参考基因拷贝数,COLLIER等[16]研究中使用双重ddPCR获得了水稻、小麦、玉米、番茄、马铃薯和柑桔类物种的高质量转基因拷贝数测量值。其中提到 ddPCR分析产生的拷贝数测量值非常接近整数值,通常与使用Southern blot杂交得出的结论相近。并且,基于ddPCR的方法在基因组非常大的物种(如小麦和玉米)中也能很好地发挥作用,但传统的DNA印迹杂交方法在这一方面却极富挑战性。因此快速准确地确定转基因拷贝数并鉴定大量样品的半合子和纯合子的能力使 ddPCR成为植物生物学研究人员的强大工具。【本研究切入点】六倍体小麦因其基因组庞大复杂(约17 Gb),利用传统Southern blot等方法检测拷贝数费时费力,无法达到高通量检测的要求,当需要对大量的转化试验进行拷贝数分析时,传统方法无法满足要求。【拟解决的关键问题】本研究以中国农业科学院作物科学研究所小麦抗逆分子育种课题组利用农杆菌介导法获得的转nib8小麦为试验材料,以小麦PINb-D1b(籽粒硬度)为参考基因[15],期望通过数字 PCR技术建立一种快速稳定的小麦基因拷贝数检测方法,为促进小麦基因组研究以及基因工程研究提供有力工具。
1 材料与方法
1.1 试验材料
2018年10月,中国农业科学院顺义基地种植转nib8小麦株系(由中国农科院作物科学研究所小麦抗逆分子育种课题组提供)和扬麦16(受体)。
1.2 内参基因和目标基因引物设计
选取位于小麦 D基因组中的单拷贝纯合基因PINb-D1b(籽粒硬度)作为试验内参基因[17],通过引物的筛选和比较,确定专一性和特异性强的引物(表1)。设计Wx012(蜡质基因)和SSII(淀粉合成酶)2个内参基因的引物和探针,确保探针的特异性(表1)。
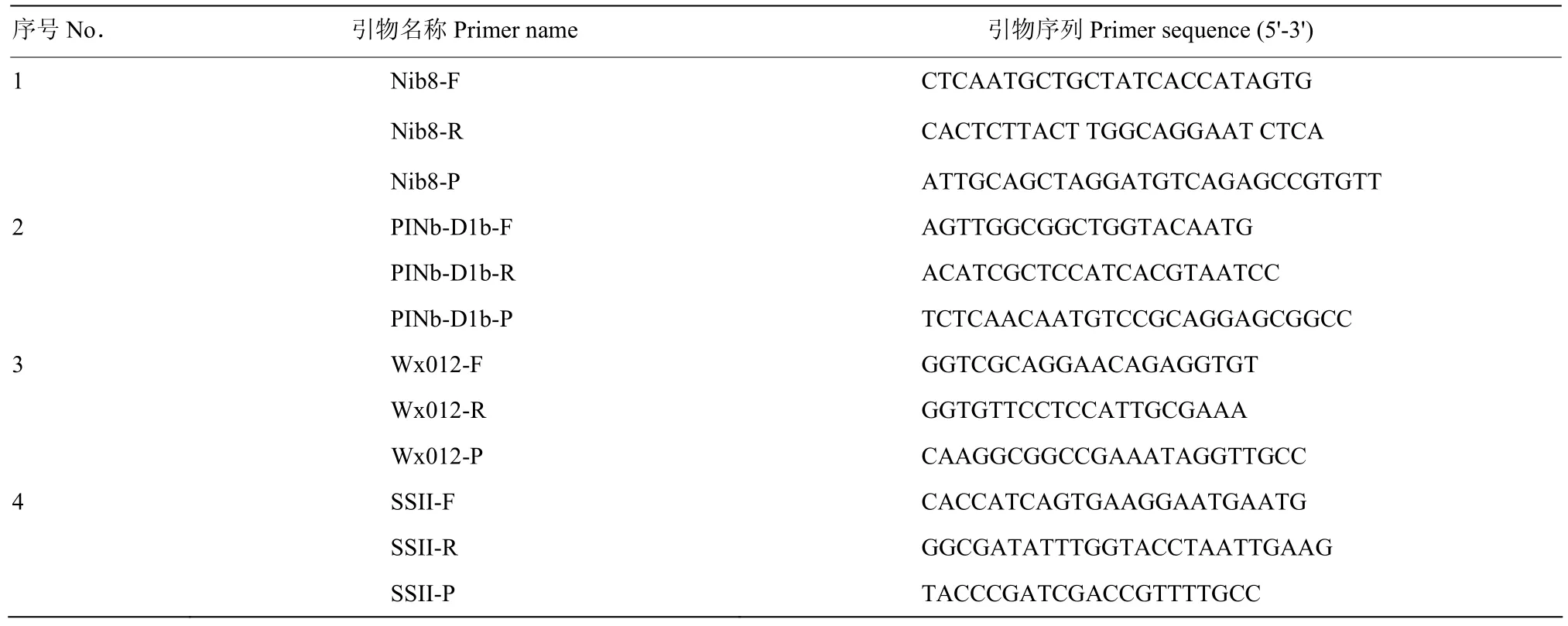
表1 数字PCR 探针引物序列Table 1 Sequences of probes and primers used in digital PCR
1.3 样本DNA的提取
结合CTAB法和全自动核酸提取仪提取法提取样本DNA[18]。
1.4 片段化基因组
用EcoRⅠ(NEB公司)对叶片基因组DNA进行核酸内切酶预处理,每微克DNA加入1个单位EcoRⅠ。37℃处理过夜,65℃ 30 min使酶失活,并将模板DNA稀释为 20 μg·μL-1。
1.5 制备反应体系及PCR反应
采用以DNA为模板的探针法。反应体系见表2,反应条件为95℃ 3 min;95℃ 30 s,57℃ 1 min,40个循环;98℃ 10 min。为保证高效的模板扩增,PCR仪的升降温速度需低于2℃·s-1。
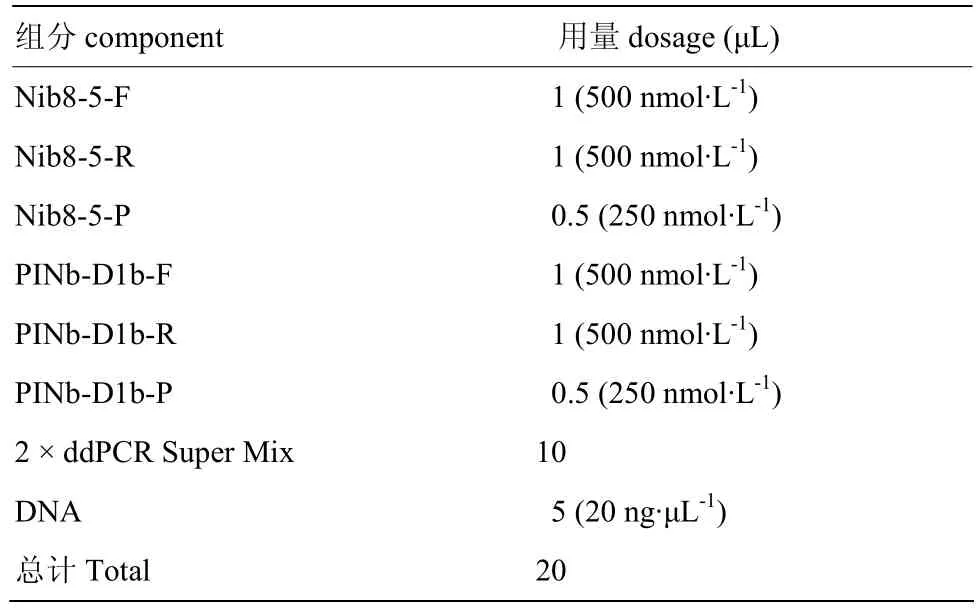
表2 PCR扩增反应体系Table 2 PCR amplification reaction system
1.6 微滴分析
将 96孔板放入微滴分析仪中,读取微滴荧光信号,从而判断微滴的阳性或阴性。使用 QuantaSoft V1.3.2软件计算每个样品目的序列的微滴数(拷贝/μL)[19],泊松分布的置信区间为 95%;然后根据内参基因的拷贝数计算待测目的基因的拷贝数。
2 结果
2.1 引物的特异性筛选
选用转nib8小麦株系为模板,使用表1的4个基因引物进行PCR扩增(图1)。结果发现扩增出的条带均为单一目的条带,表明引物具有特异性。
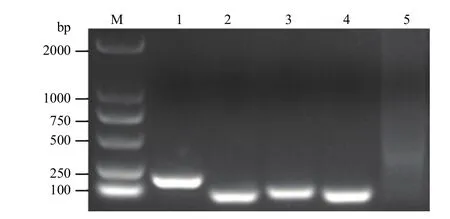
图1 4对引物的特异性扩增Fig. 1 Specific amplification results of four pairs of primers
2.2 退火温度的筛选
通过对退火温度的梯度分析,在6个温度条件下,nib8均表现出良好的扩增图形,且随着退火温度的升高,nib8的阴性微滴和阳性微滴区分的愈发明显,扩增越来越特异(图2-A)。图2-B中内参基因随着退火温度的升高,扩增效率变化不明显。因此,为使nib8和内参基因均能表现出良好的扩增效果,最终以59℃作为最优退火温度。
2.3 灵敏度(检测低限)和准确性检测
2.3.1 DNA模板浓度的筛选 由图3可知,nib8在4个不同模板浓度下均扩增出良好的微滴分离现象,随着浓度的增加,微滴数增加。然而,nib8在160 ng模板浓度时,阳性微滴和阴性微滴已无法明显分开,在320 ng模板浓度时,该现象愈发明显。模板浓度越高,体系扩增效果越差,微滴不能明显分开,而模板为20 ng时的微滴数较少,无法明显看出扩增效果,因此,最终选定40 ng为最佳模板浓度。
2.3.2 特异性检测 使用QuantaSoft V1.3.2软件对6个转nib8小麦株系的拷贝数进行分析。由图4可以看出阳性微滴数和阴性微滴数区分明显,说明体系稳定。由表3可以看出数字PCR检测结果中有3个株系为1个拷贝,可以用于进一步基因功能分析。
2.4 体系验证及应用
2.4.1 不同检测方法的比较 由表 4中可以看出数字 PCR的检测结果与通过染料法和探针法进行的real-time PCR分析结果一致。由图5可以看出转nib8小麦第16号株系的Southern blot结果也与数字PCR结果一致,均是1个拷贝。
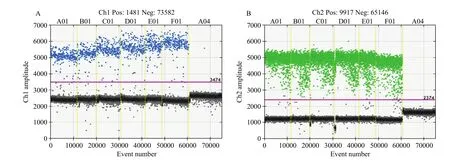
图2 不同退火温度条件下nib8(A)和内参基因(B)的数字PCR检测Fig. 2 Digital PCR detection of nib8 (A) and internal reference genes (B) at different annealing temperatures

图3 不同模板浓度条件下nib8(A)和内参基因(B)的数字PCR检测Fig. 3 Digital PCR results of nib8 gene(A) and control gene (B) at different template concentrations
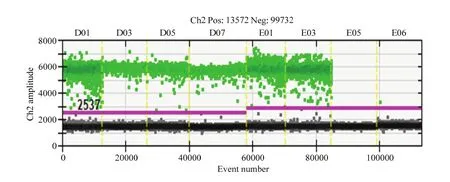
图4 转nib8株系特异性检测Fig. 4 Specific detection of nib8 strains was performed
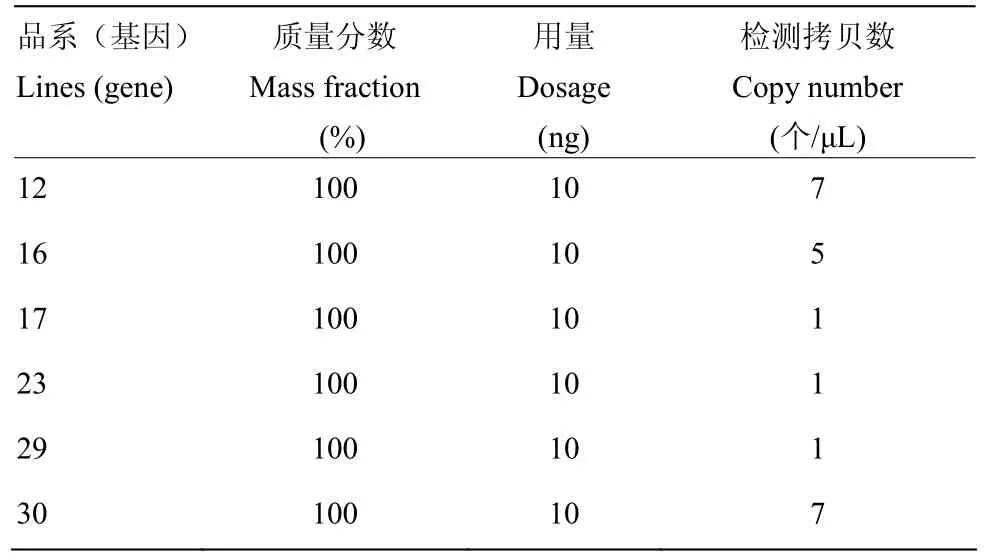
表3 转nib8株系的数字PCR检测低限测定结果Table 3 The results of low limit determination by digital PCR were obtained by transplanting nib8 strain
2.4.2 内参基因的选择 在内参基因的选择上,除小麦D基因组中的单拷贝纯合基因(PINb-D1b)外,同时还开发了其他小麦内源基因作为拷贝数检测的内参基因。
以Wx012和SSII为内参基因检测部分转nib8小麦株系目标基因的拷贝数(图6和图7),其结果与用PINb-D1b为内参基因的分析结果一致,都是1个拷贝,证明这3个基因都可作为内参基因用于小麦目标基因拷贝数数字PCR分析。
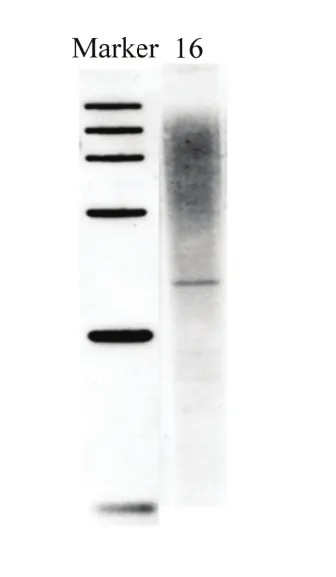
图5 转nib8小麦第16号株系的Southern blot检测Fig. 5 Southern blot results of nib8 transgenic wheat line 16
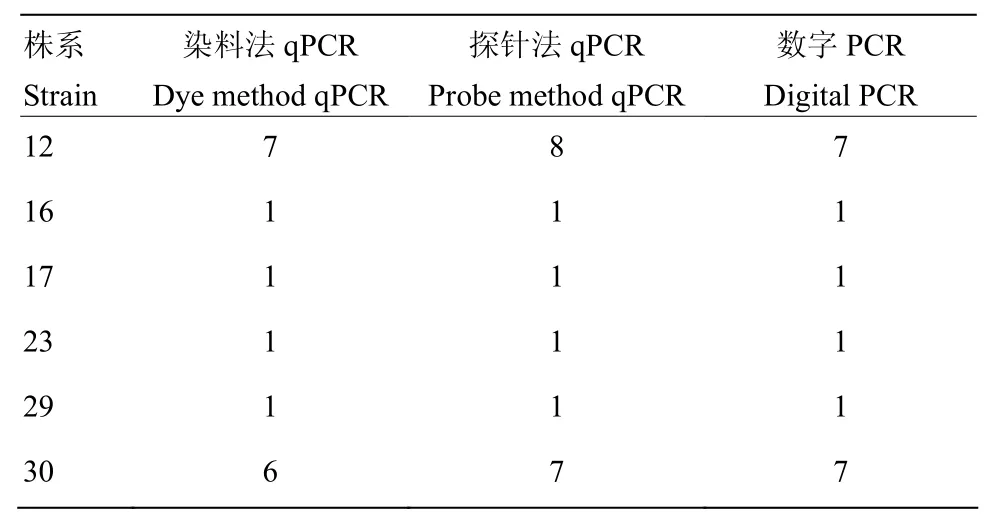
表4 探针法、染料法和数字PCR的结果对比Table 4 Comparison between fluorescence quantitative method such as probe method and dye method, and digital PCR method
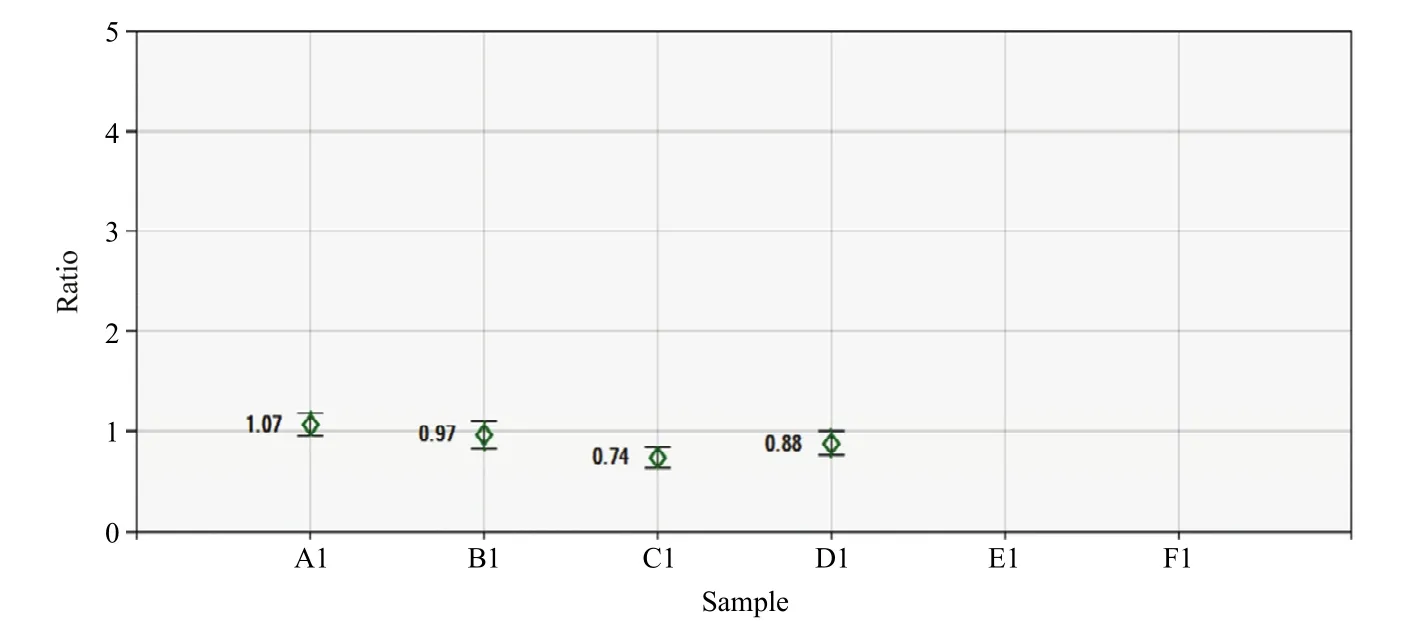
图6 内参基因的拷贝数分析Fig. 6 Copy number analysis of internal reference genes
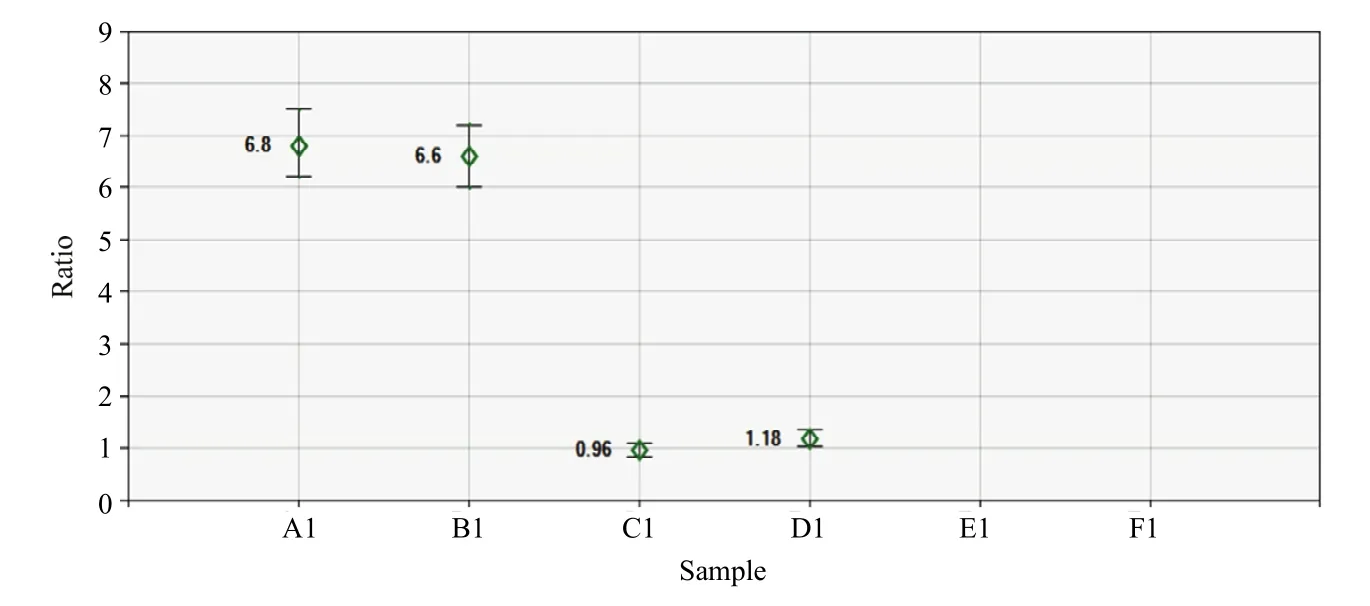
图7 转nib8株系的拷贝数分析Fig.7 Copy number analysis of transferred nib8 lines
2.4.3 靶基因不同区段的检测效果验证 在nib8的不同区段进行引物探针设计,分别以PINb-D1b、Wx012和SSII为内参基因检测nib8拷贝数,结果均一致(图8-A,图8-B),第12号转nib8小麦株系的拷贝数都是7(图8-C)。
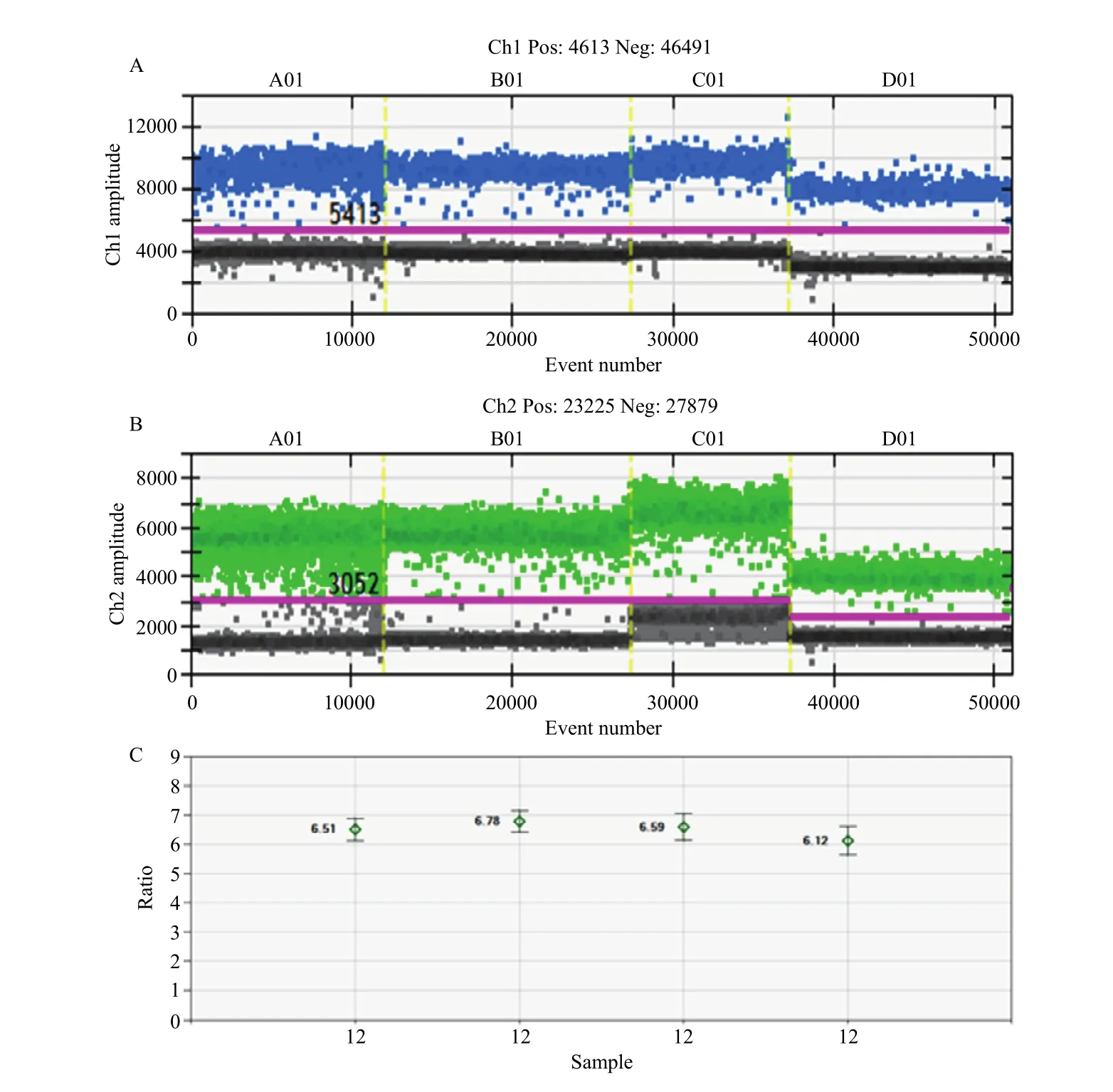
图8 在nib8不同位置设计引物的结果图(第12号转nib8株系)Fig.8 Results of designing primers at different positions of nib8 gene (take 12 strains as an example)
3 讨论
3.1 不同拷贝数检测方法的比较
目前,小麦拷贝数检测方法很多,但用数字PCR检测转基因小麦外源基因拷贝数报道较少。经典的外源基因拷贝数检测方法主要有 qRT-PCR和 Southern blot。其中qRT-PCR分析方法的步骤较为繁琐,首先需要准备标准 DNA样品,然后利用此样品构建标准曲线,而标准曲线的建立又涉及体系的摸索与优化,需要耗费大量的时间和精力,所需试验周期较长[20];另外,借助标准曲线进行定量本身就是一种相对定量方法,所以试验结果并不准确[21]。而传统的Southern blot方法不但对DNA需求量大,试验操作步骤复杂,而且对人员操作的技术要求比较高[22]。与定量PCR不同,数字PCR无需标准曲线即可实现对DNA样品绝对定量的灵敏、准确。一个PCR反应被分成许多独立的反应,每个反应都有一个阳性或阴性的信号。应用泊松统计量,直接从阳性反应和阴性反应的数量计算出原始样品中DNA分子的数量。本研究中数字PCR模板要求低,而且相比Real-time PCR的灵敏性(0.1 pg·μL-1)高,数字 PCR 灵敏性能达到 0.001 pg·μL-1,是Real-time PCR的100多倍。相比Southern blot,数字PCR试验周期短,操作简单,检测通量高。
3.2 体系准确性的比较
一步多重PCR(multiplex PCR)又称多重引物PCR或复合PCR,是指在同一PCR反应体系中添加2对以上引物,同时扩增出多个核酸片段的PCR反应,且每对引物只对应1个[23]靶标片段,与常用的RAPD技术(1对引物对应多个扩增产物)[24]全然不同。考虑到二重微滴式数字PCR不仅能避免单重ddPCR定量因二次取样所产生的样品内的误差,而且可以消除不同样品 DNA因取样不一致而造成的样品间的误差,所以本研究采用二重PCR反应扩增,选用了3个不同的小麦单拷贝基因Wx012、PIN-b1和SSII作内参基因,其中以PIN-b1为内参基因时,Wx012和SSII的拷贝数为1(图6)。又以这3个基因为内参基因检测转基因株系12的拷贝数时,结果也相同。为了检验表达是否一致,在nib8序列的不同区段设计引物,结果显示一致。为了试验结果的准确性,本研究选择小麦基因组里的其他2个单拷贝基因Wx012和SSII作为内参基因检测PIN-b1[25]。以此来检测该体系的准确性。
3.3 数字PCR检测小麦拷贝数方法的应用
本研究建立的检测体系短时间内可以检测几百个样品,大大提高了工作效率。本研究建立的拷贝数方法不仅可以检测外源基因。也可以检测内源基因的拷贝数。因此,本方法具有快速、灵活、高效、所需样本量少等优点,而且已经在基因表达研究、基因组拷贝数鉴定、癌症标志物稀有突变检测、转基因成分鉴定等诸多领域显示出广阔的应用前景[26-28]。BURNS等[29]应用数字 PCR来评估几种转基因分析方法的检测限。
DOBNIK等[30]应用数字 PCR技术,针对欧盟授权的 12种转基因玉米品系建立了转基因成分多重定量分析方法。目前,该技术已经快速应用于各个领域。
4 结论
建立了针对小麦双重微滴式数字 PCR检测基因拷贝数的方法。确定了引物和探针的最佳终浓度分别为500和250 nmol·L-1,退火温度为59℃。该检测方法可以选用3个内参基因(Wx012、PIN-b1和SSII)。
