B2-AgMg金属间化合物点缺陷行为的理论研究
2020-05-13郝志强姚建刚尹登峰
郝志强, 李 辉, 姚建刚, 尹登峰,2
(1. 烟台南山学院 工学院,烟台265713; 2. 中南大学材料科学与工程学院,长沙410083)
1Introduction
With the trials of lower density and higher specific strength, specific stiff, Mg-based alloy plays an important role in the fields of electronic, automobile and aviation industries as the lightest constructional material[1-4]. However, the restrained mechanical properties limit its use to a certain extent[5]. Alloying is an effective method to improve mechanical properties of Mg-based alloys, of which the precipitated intermetallic compounds significantly contribute to this improvement . In light of this fact, much attention focused on the research of thermodynamic properties of various intermetallic compounds in Mg-Al-Ca, Mg-Sn-Mn, Mg-Zn-Mn, Mg-Zn-Caetalternary magnesium alloys[6-12]. Such studies have been of practical significance to develop new product and alloy design. Earlier studies have shown that many precipitations are off-stoichiometric in a narrow range[13], and this off-stoichiometry is related to the arise of native point defects of the ordered intermetallic compounds. So, the investigation of the behaviors of point defects for intermetallics Mg2Ca, MgZn2, MgCu2, and Mg24Y5were performed in recent years[14-17], which mainly focuses on the varying of the electronic properties of the defective intermetallic compounds.
By contrast, the exploration of the Ag-Mg-Zn ternary magnesium alloy appears to be limited, and the recent research is about the structural stability, elastic and thermodynamic properties of the intermetallic compoundsMgAg, Mg4Zn8and Ag8Mg4Zn4under high pressure and high temperature[18]. Up to now, no any report related to the elastic properties for these intermetallics with point defects in Ag-Mg-Zn ternary magnesium alloy could be found.
As we know, the compositional off-stoichiometry with point defects affects the mechanical and physical properties of intermetallic compounds. However, investigating this effect for a precipitate phase of Ag-Mg-Zn alloy appears to be difficult experimentally . Therefore, further theoretical studies are necessary to explore this exhibition.
In this work, intermetallic B2-AgMg was chosen as subject, and a series of theoretical studies were fulfilled to explore the thermodynamic properties for the defective B2-AgMg. According to Ref.[14-17], four kinds of native defects are produced in intermetallics, namely Mg vacancy (MgV), Ag vacancy (AgV), Mg anti-site (MgAg) and Ag anti-site (AgMg). Here, we predict the possible changes of elastic and thermodynamic properties of B2-AgMg caused by the formation of different point defects, this is different from previous studies in regard to the point defects behaviors.
2Computational methodology
Present works were performed by using the density functional theory (DFT) code VASP (Vienna Ab-initio Simulation Package)[19]. The ion-electron interaction was modeled with the projector augmented wave (PAW) potentials[20].The exchange-correlation energy by the generalized gradient approximation (GGA) of Perdew was employed for all elements in our models by adopting Perdew-Burke-Ernzerh (PBE) of parameters[21]. All of the calculations applies a sufficient cutoff energy of 350 eV for plane waves basis sets. The point defect calculations used a 4×4×4 (128 atoms) supercell of BCC B2-AgMg and a 4×4×4 Monkhorst-Pack k-mesh for Brillouin-zone integration. All ground-state configurations were obtained by allowing full relaxation on both volume and shape and by minimizing the Hellman-Feyman forces until the total force on each ion were converged within 0.02 eV/Å.
3Results and discussion
Validation of the parameter settings was first carried out by predicting structural parameters and elastic modulus of bulk Ag, Mg, and B2-AgMg. The calculated results (Ag-4.16Å, Mg-3.21Å, and AgMg-3.336Å) are in agreement with the experimental values (Ag-4.09Å,Mg-3.209Å, and AgMg-3.311)[22-23]. Also, the calculated bulk modulus (63.18GPa) for B2-AgMg agrees well with other theoretical results using the same pseudopotential (65.1GPa)[18]. Obviously, the method we used here is appropriate to performe this project.
3.1Electronic properties of pure B2-AgMg
First, electron local function (ELF) and total density of states (DOS) of perfect super crystal for B2-AgMg were calculated to discuss the electronic properties. As shown in Tables 1-c, the value of electron local function is 0.2 in the main region of (0 1 1) plane, indicating that strong metallic bonds plays a leading role in perfect super crystal. However, covalent interactions between all of the Mg atoms are also observed, similar with Mg2Ca and Mg24Y5[14, 16]. This can be further verified by the total density of states (Tables 1-d). One can see that typical metallic peaks are mainly distributed in the bonding area (-1.5eV-0), and a few electronic distribution is observed in the anti-bond domain (0-0.2eV). The feature of pseudogap is not more evident, suggesting that the dominant role between all atoms is not the covalent bonding. This is in agreement with the electron local function.
3.2Stability of defectiveB2-AgMg
Different types of point defects, as shown in Fig.2, were introduced into the perfect lattice with a 128-atomsupercell.
The thermodynamic stability of corresponding defectiveAgxMgycan be measured by the formation energy which is defined as Eq. (1).To evaluate the relative concentrations of different point defects, we assess the defect formation energies (ΔE) for MgV, MgAg, AgV, and AgMgwith Eq. (2) and (3).
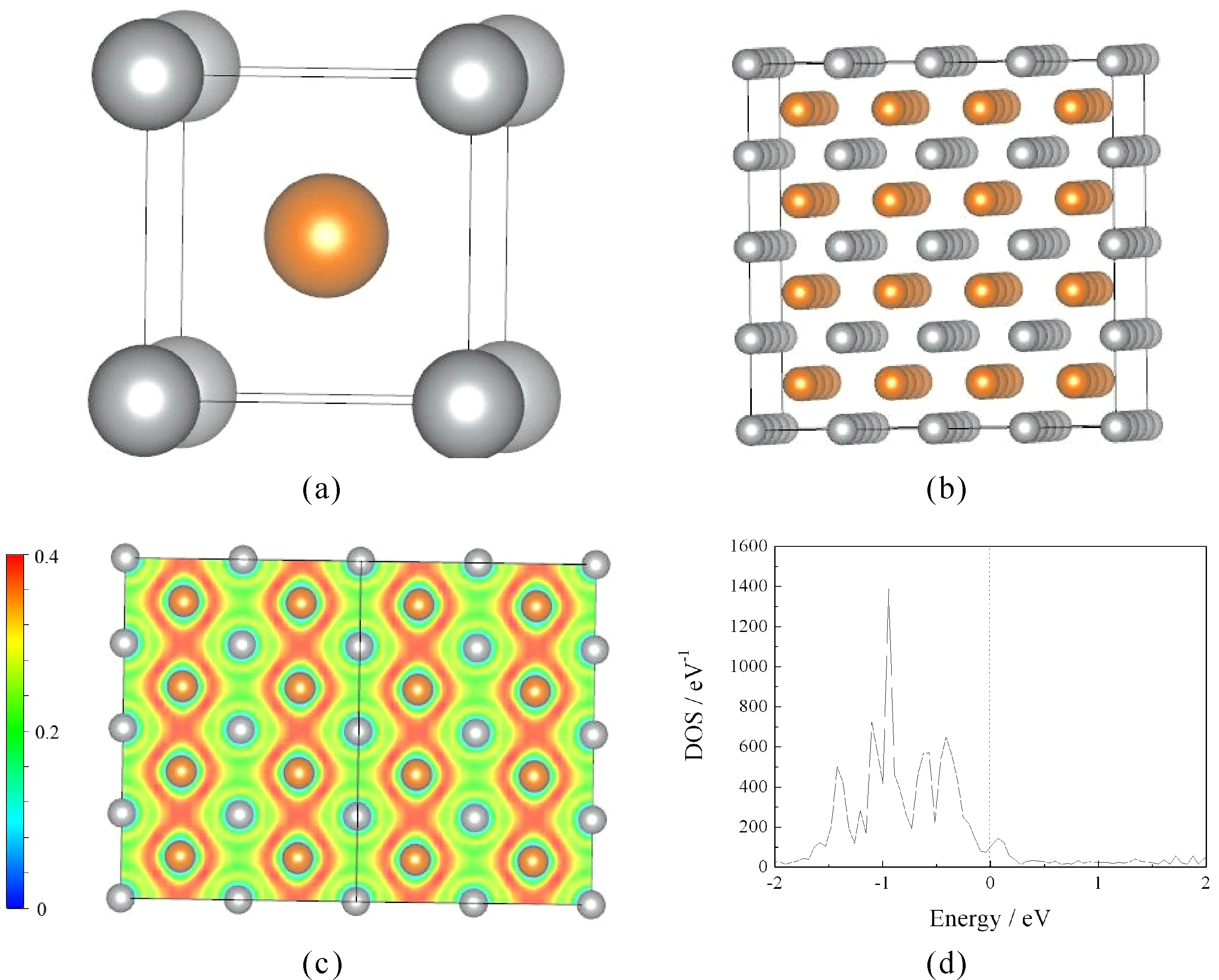
Fig.1. Crystal model (a), perfect super crystal (b), electron local function on ( 0 1 1) plane (c), and total density of states (d) of perfect super crystal of B2-AgMg. (Yellow and gray balls represent Mg and Ag atoms, respectively).
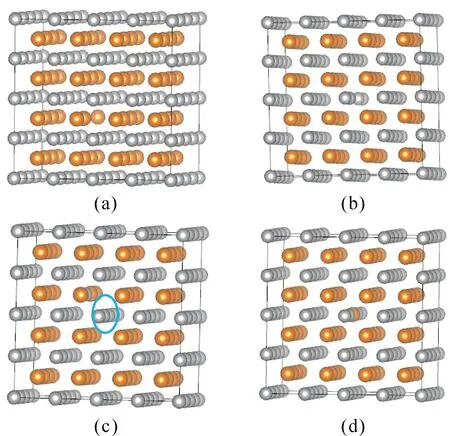
Fig.2 Supercell models for point defect calculations of B2 -AgMg intermetallic (The yellow and grey balls represent Mg and Ag atoms, respectively.a:Mg vacancy b: Ag vacancy c: Mg anti-site d: Ag anti-site)
(1)
(2)
(3)
whereEAgxMgyandEAg/Mgare the total energy of the defectiveAgxMgysupercell, and the atomic energy of Ag and Mg in each pure bulk, respectively.Epis the total energy of perfect B2-AgMg (4×4×4) supercell, andEtrepresents the total energy of B2-AgMg (4×4×4) supercell with one single vacancy or anti-site.
All calculated results are displayed in Table 1.It is evident that all theEfvalues are negative, implying that all the defective supercells can be produced favorably in Ag-Mg-Zn alloys concerning the energy. Also, bothEfandΔEhave relatively smaller value for MgAgand AgMgthan those of MgVand AgV, which suggests that the anti-site defects are easier to be formed than those of vacancy defects.
As known to all,the appearance of point defects can form the localized states, and the wave functions of localized states will interact with around atoms to make lattice atoms away from the equilibrium position.Aiming at giving a physical interpretation, we calculated the electron local function of all B2-AgMg supercells.
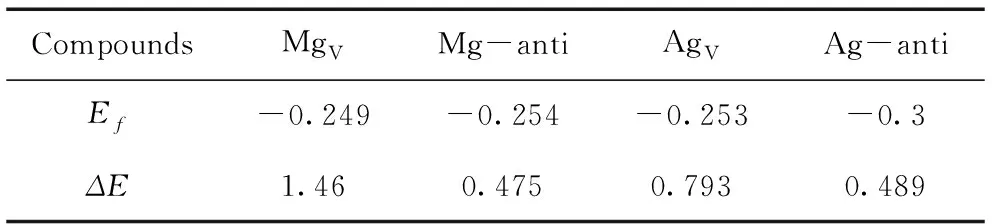
Table 1 Lattice formation energy Ef(eV) and formation energy of point defects ΔE(eV).
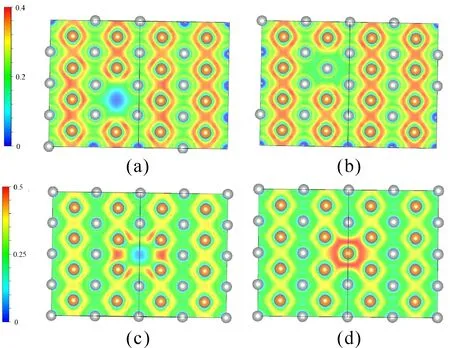
Fig.3 Electron Local Function of correspondingAgMg supercell on (1 1 0 ) plane: (a) Mg vacancy, (b) Mg anti-site, (c) Ag vacancy, (d) Ag anti-site.
It is found in Fig.3 thatelectron distribution decrease obviously around Mg vacancy site because of the emerge of Mg vacancy [Fig. 3(a)], whereas clear localized states of electrons is observed in [Fig. 3 (c)], showing that Ag vacancy contributes to affect the covalent bonding between Mg atoms.
Different fromvacancy defects, Mg anti-site point defects has no evident effects on the electron distribution [Fig. 3 (b)], but the distinct varying for the electron distribution is the appearance of covalent bond between Ag anti-site point defects and the surrounding atoms [Fig. 3 (d)].
It is noted that all changes of bonding properties are mainly around the site of point defects, and no evident variation can be observed in other region.
To gain morevaluable information about the electronic structure for B2-AgMg with point defect, the total DOS of them were calculated which are plotted in Fig.4. It is seen that for vacancy defects (Fig. 4-a and 4-c), Fermi level shifts to the right, especially for Agv, leading to the higher value of DOS at the Fermi level in comparison with the DOS of perfect B2-AgMg (Fig. 3-d). Clearly, higher amount of density just at the Fermi level means lower stability of corresponding crystal, in agreement with the analysis of defect formation energies. By contrast, anti-sits defects, particularly AgMg exhibits more evident characteristics of pseudogap, so it is more possible to has better stability. However, we also found that more electrons occupied the anti-bond states of all defective B2-AgMg supercell, so the structural stability for them seems to be wose compared to perfect B2-AgMg supercell

Fig.4 Total densities of states of defective B2-AgMg supercell : (a) Mg vacancy, (b) Mg anti-site, (c) Ag vacancy, (d) Ag anti-site.
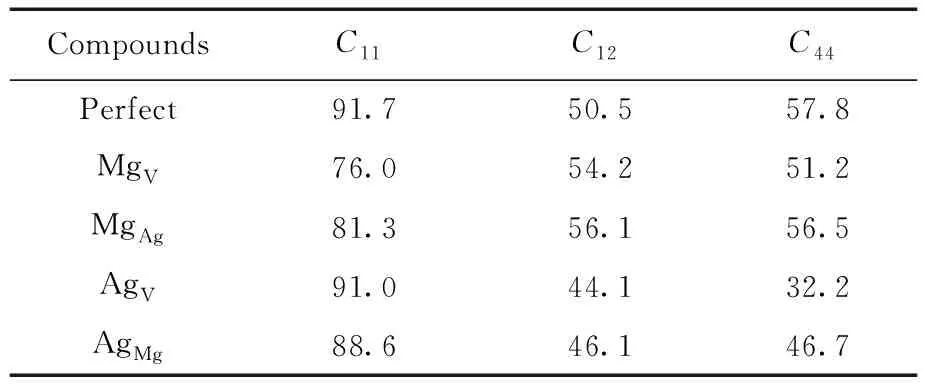
Table 2 The elastic constant(GPa) of pure and defective B2-AgMg
3.3Hardness of B2-AgMg intermetallics with point defects
For a given crystal, thermodynamic properties are related to the elastic constants of corresponding crystal[24], so the elastic constants, as shown in Table 2, for MgV, AgAl, MgAg, and AgMgwas calculated to predict their effects on elastic properties.
The bulk modulusB0, shear modulusG, Youngs modulusE, and Poisson ratioνis defined as the following equations:
(4)
(5)
(6)
(7)
According to Ref. [24], hardness of crystal is closely associated with the shear modulusGand Youngs modulusE, and largeGandEalways suggest the high hardness of corresponding crystal. Also, there is a linear connection betweenC44and thehardness. Crystal with high hardness generally has the largeC44.
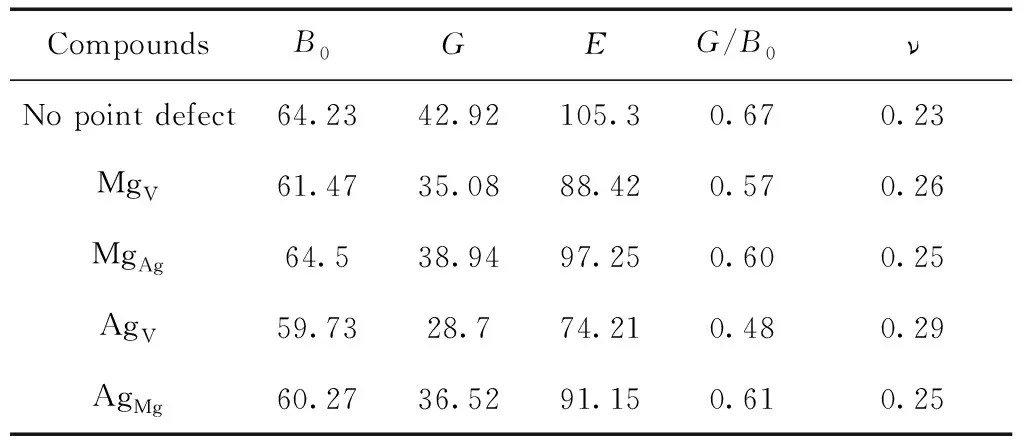
Table 3 The Bulk modulusB0(GPa), Youngs modulusE(GPa), Shear modulus G(GPa) of pure and defective B2-AgMg
From Table 2 and 3, we can learn that the value of shear modulusGand Youngs modulusEof B2-AgMg with these four point defects decrease significantly compared to the pure B2-AgMg, implying that the appearance of point defects tends to attenuate the hardness of B2-AgMg compounds. This is particularly apparent for AgV, of which the reduced value reaches 14.22 GPa. Also, equivalent conclusion can also be drew from the variation of elastic constantC44.
3.4Brittleness of B2-AgMg intermetallics with point defects
Ductility and brittleness of materials can be evaluated by the rule of thumb for Pugh[25], and the material with large value ofG/B0generally has the characteristic of brittleness, while it exhibits the ductility ifG/B0is small. According to this criterion, we found that the brittleness for B2-AgMg compounds with these four point defects decreases slightly because of the reduced value ( from 0.06 to 0.19 ) ofG/B0.
From Pugh criterion, we also know that ifG/B0is more than 0.57, corresponding material is brittle, or it is ductility. Our calculated value ofG/B0(0.67) indicates that B2-AgMg compounds is brittle, in agreement with the earlier report[18]. In fact, the appearance of point defects, except for AgVwhich appears to be ductility, has nothing to do but abate the effects on brittleness to varying degrees, and the nature of brittleness for B2-AgMg is not essentially changed. This is similarly predicated by Poisson ratiov, the value ofvless than 1/3 is customarily set down as the brittle material.
3.5Thermal properties of B2 -AgMg intermetallics with point defects
Debye temperature can be applied to reveal the interparticle potential between atoms of corresponding crystal. Herein, we calculated the Debye temperature based on elastic constant evaluations, and it could be obtained by the following expression:
(8)
Wherehis the Planck constant,NAis the Avogadro number,Mis the molecular weight andρis the density. The average sound velocityvmis expressed as follows:
(9)
(10)
(11)
(12)
whereB0andGare isothermal bulk modulus and shear modulus,vlandvsrepresent the compressional velocity and shear sound velocity, respectively.

Table 4 Debye temperature ΘD(K) and average sound velocity vm(m/s) of pure and defective B2-AgMg
Predicated Debye temperature forpure and defective B2-AgMg intermetallics are given in Table 4, and this value for pure B2-AgMg is 331.6 K. What needs to be explained is that in Ref. [18], Luetalcalculated this value with 600K. We think that their works in this regard seem not to be correct enough. In general, high melting point for a given intermetallic compound always means the comparatively high Debye temperature. However, they come to a contrary conclusion in comparing the connection between melting point and Debye temperature for AgMg, Mg4Zn8,and Ag8Mg4Zn4complexes. As they do, B2-AgMg has a low Debye temperature, but the highest melting point and the most stable thermodynamic properties compared to Mg4Zn8and Ag8Mg4Zn4. In addition, Taoetalalso calculated the Debye temperature for the same structure of B2-MgR (R=Sc, Y, La, Ceetal) binary complexes, and they predicted the largest value for Debye temperature in B2-MgR series is merely 375K of MgSc[26], and this value decreases in sequence as the increase of the molecular weight. Apparently, the Debye temperature of B2-MgAg should not exceed 375 K of MgSc due to its large molecular weight, and this is unanimous with our calculated result.
As we know, higher Debye temperature is the reflection of the strongerinteractions between all atoms in crystal. According to the outcomes, we found that all of the point defects in B2-AgMg contribute to decrease such interactions, and this is particularly significant for AgV, of which the extent of reduction is about 20%.
In general, the stability ofintermetallic B2-AgMg is based on its ordered structure, where the metallic bonding plays a leading role in keeping the stability of the crystal. However, the arise of point defects is incline to altering this charge population to a certain degree, giving rise to an elevation of Coulomb energy[27]. As a result, corresponding crystal exhibits lower Debye temperature.
4Conclusions
The point defects of Mg and Ag anti-site are easily produced in B2-AgMg due to the comparatively small defect formation energies. All of the point defects contribute to decrease the hardness, brittleness and Debye temperature of corresponding B2-AgMg crystal.
