CRISPR/Cas9系统中的脱靶效应及检测技术研究进展
2020-03-19张晨雷展李凯商颖许文涛
张晨 雷展 李凯 商颖 许文涛
(1. 昆明理工大学农业与食品学院,昆明 650504;2. 中国农业大学食品科学与营养工程学院 北京食品营养与人类健康高精尖创新中心,北京 100083;3. 农业部农业转基因生物安全评价(食用)重点实验室,北京 100083)
CRISPR/Cas9系统是细菌和古生菌免疫病毒或噬菌体侵害的一种获得性免疫机制,其中sgRNA可特异性识别外源遗传物质,Cas9蛋白可靶向切割双链DNA。因为组成简单、特异性好、切割效率高,CRISPR/Cas9系统经改造后成为新一代基因编辑工具并迅速被广泛应用。在实际应用过程中,CRISPR/Cas9系统优势凸显,其快速、准确的基因编辑能力,使通过基因定点突变治疗人类遗传疾病成为可能。然而在基因编辑过程中存在的脱靶效应,严重阻碍了CRISPR/Cas9系统的发展。因此,为明确CRISPR/Cas9系统产生的脱靶效应及提高基因编辑效率,本文综述了CRISPR/Cas9系统的脱靶类型、影响因素、降低策略以及脱靶检测技术的最新研究进展。
1 CRISPR/Cas9系统简述
1987年,在研究大肠杆菌碱性磷酸酶(iap)基因功能时,首次发现了串联间隔重复序列[1]。2002年,这种结构被命名为聚类规则间隔的短回文重复序列,临近的一组基因被命名为Cas[2]。2007年,证实CRISPR及Cas编码的蛋白与细菌的获得性免疫机制有关[3]。2013年,首次利用CRISPR/Cas9系统实现对人和小鼠内源基因的高效编辑[4-5]。
在自然界中,约90%古生菌和40%细菌通过基因组或质粒上存在的CRISPR/Cas系统对外来病毒或噬菌体的侵害进行免疫。当病毒或噬菌体首次将遗传物质注入细菌内部时,Cas蛋白会将外源遗传物质上的一小段序列整合到CRISPR序列的5'端,形成对外源遗传物质的“免疫记忆”。当细菌再次遭遇侵害时,含有外源遗传物质信息的CRISPR序列转录形成sgRNA。sgRNA与Cas蛋白结合,从而特异性切割外源DNA。外源DNA被切断后沉默,细菌对外来病毒或噬菌体“免疫成功”[6]。
CRISPR/Cas系统存在多种类型,依据其编码的效应蛋白可以将其分为两大类(图1)。第1大类CRISPR/Cas系统的效应蛋白是由含有4-7个Cas蛋白组成的多亚基效应复合物组成。第2大类CRISPR/Cas系统中仅包含单一的多结构域效应蛋白。两大类系统又被细分为6种类型,其中第1大类包含类型I,III和IV,第2大类包含类型II,V和VI。根据特征蛋白的不同,可区分类型I、II、III。其中Cas3蛋白代表类型I,Cas9蛋白代表类型II,Cas10蛋白代表类型III。类型IV缺乏由Cas1和Cas2蛋白组成的适应模块;类型V具有预测效应蛋白Cas12a;类型VI是唯一具有毒素特性的靶向RNA的CRISPR/Cas系统[7]。因为效应复合物结构简单,第2大类CRISPR/Cas系统被改造成为基因编辑工具。其中类型II的CRISPR/Cas9系统是最常见且广泛应用于各个领域的基因编辑工具[8]。

图1 CRISPR/Cas系统分类图[8]
CRISPR/Cas9系统由Cas9蛋白和sgRNA两部分组成。Cas9含有HNH和RuvC核酸酶结构域,其中HNH结构域切割互补DNA链,RuvC结构域负责切割非互补链。sgRNA由CRISPR RNA(crRNA)和反式激活RNA(tracrRNA)组成,可识别约20 nt的靶序列。在基因编辑时,Cas9首先与sgRNA形成复合体;然后由Cas9识别特定的前间隔序列邻近基 序(Protospacer adjacent motif,PAM);sgRNA与靶序列特异性结合,Cas9切割双链DNA,产生双链DNA断裂(Double-strand DNA breaks,DSB)。最后,通过细胞内部的非同源末端连接(Non-homologous end joining,NHEJ)或同源重组机制(Homologous recombination,HR)对断裂的DNA引入插入缺失、修复或替换(图 2)[9]。

图2 CRISPR/Cas9作用机制及DNA自我修复过程图[10]
第一代基因编辑技术因为涉及到HR,效率低下且耗时费力。第二代基因编辑技术ZFN和TALEN的出现,基因编辑效率大幅提高。ZFN和TALEN均是合成蛋白,对靶序列切割后可产生黏性突出端DSB。ZFN和TALEN大大加快了基因编辑技术的发展,但在应用过程中,其复杂的蛋白设计、昂贵的成本和较高的难度,仍使基因编辑技术的广泛应用受限[11]。
在此背景下,第三代基因编辑技术CRISPR/Cas9系统应运而生。同ZFN和TALEN相比,CRISPR/Cas9系统不需要设计复杂的DNA结合蛋白以及DNA结合蛋白与Fok I核酸酶的融合过程。通过软件就可快速设计sgRNA,并对其进行初步筛选。同时,通过改变sgRNA中的一小段序列,CRISPR/Cas9系统可快速实现对其他基因位点的编辑。由于过程简单,成本低廉,规模化,高通量,编辑效率高,实验要求低等特点,CRISPR/Cas9系统迅速被应用到广泛的研究领域当中[11]。例如医学领域,CRISPR/Cas9系统已经建立了多种细胞系及动物模型[12];动物科学领域,利用CRISPR/Cas9系统生产更广泛的基因修改动物[13];植物科学领域,CRISPR/Cas9系统促进作物育种,加速作物改良,增强全球粮食安全等[14]。
2 CRISPR/Cas9系统的脱靶类型
CRISPR/Cas9系统中的脱靶突变主要是由于在复杂的基因组中sgRNA存在数量庞大的脱靶位点所造成的。此外,Cas9识别低频率的“NAG”模式的PAM序列也会导致脱靶,但PAM序列对错配容忍度较低[15]。现阶段,根据连续碱基不同错配、间隔碱基不同错配以及PAM近端远端不同错配可以将CRISPR/Cas9系统的脱靶类型简单分成3类。
对于连续碱基不同错配,Fu等[16]基于定量人类细胞的增强型绿色荧光蛋白(Enhanced green fluorescent protein,EGFP) 破 坏 试 验, 评 估 了CRISPR/Cas9系统中sgRNA与DNA界面内连续不同错配对Cas9活性的影响。结果表明,对于单碱基错配,sgRNA的5'末端错配率大于3'末端。但是在sgRNA的3'末端,也会存在一些位置对错配具有良好的耐受性,并且不同靶序列对sgRNA错配敏感的特定位置是不同的。双重错配实验显示出类似于单碱基错配的实验结果,而引入3个或更多个错配则会导致Cas9核酸酶活性显著丧失。
对于间隔碱基不同错配,同样基于定量人类细胞的EGFP破坏试验,Fu等[16]对不同位置间隔的双重错配进行了表征。不同位置间隔的双重错配实验显示出类似于单碱基错配的实验结果,即sgRNA的5'末端错配率大于3'末端。
对于PAM近端远端不同错配,之前的研究表明,靶序列上与PAM序列紧邻的8-12个碱基决定CRISPR/Cas9系统的切割准确性。这一区域也被形象的称之为种子区域。单碱基错配敏感的特定位置集中在靠近PAM序列的5-6碱基,且不同靶标对错配敏感的位置不同。双重或多个核苷酸错配,在靠近PAM序列的5-6碱基区域同样表现出了对错配更高的敏感性,且连续错配比间隔错配更敏感。这表明靶序列中更靠近PAM序列的5-6个碱基才是决定特异性结合的关键因素。此外,对于单一或双重错配,即使在靠近PAM序列的5-6碱基区域,有时也会表现出对错配的良好耐受性[17]。
总之,靶序列对sgRNA的3'末端错配更敏感,且其特异性是复杂的和靶标依赖性的,同时在sgRNA的3'端发生单一或双重错配时,通常是良好耐受的。此外,并非所有5'端的sgRNA/DNA界面错配都具有良好的耐受性。最近,针对CRISPR/Cas9系统的脱靶,Lin等[18]提出了一种新型脱靶类型“DNA凸起”和“RNA凸起”,如图3。当脱靶位点较靶序列存在一个或多个额外的碱基(插入),sgRNA与该脱靶位点结合时,脱靶位点上多余的碱基就会形成“DNA凸起”。当脱靶位点较靶序列缺少一个或多个碱基(缺失),sgRNA与该脱靶位点结合时,sgRNA上多余的碱基就会形成“RNA凸起”。通过去除或添加sgRNA上所有可能位置上的单碱基,Lin等[18]模拟了潜在脱靶的单碱基插入或缺失的位点。结果显示,Cas9在3个区域的靶位点耐受DNA凸起:距离PAM的第7个碱基,sgRNA的5'末端和3'末端;邻近PAM序列的sgRNA凸起禁止切割。2-5 bp的DNA凸起实验结果与单碱基DNA凸起的结果类似;大于2 bp的sgRNA凸起比同等大小的DNA凸起耐受性更好,长于5 bp的sgRNA凸起未发现切割活性。
3 脱靶影响因素
虽然CRISPR/Cas9系统已经在多个研究领域中取得了优异成果,但是在实际应用过程中依然存在潜在的脱靶风险。Fu等[16]基于人类细胞实验表征了CRISPR/Cas9系统的脱靶效应,结果显示CRISPR/Cas9系统的脱靶率高达66%。为降低CRISPR/Cas9系统的脱靶效应实现其更广泛的应用,研究人员探究了影响脱靶效应的相关因素。
3.1 PAM序列
PAM序列是区分靶序列与其他DNA序列、位于靶序列3'端、高度保守的一小段序列,其长度一般为2-5 nt。PAM序列存在多种模式,如“NAG”、“NGA”等。不同模式PAM序列的CRISPR/Cas9系统切割效率不同。在CRISPR/Cas9系统中,源自化脓性链球菌的SpCas9蛋白PAM序列为典型的“NGG”模式。其基因切割效率最高,N代表任意一种核苷酸,如“AGG”、“CGG”等。在基因编辑过程中,Cas9首先识别PAM序列,待sgRNA与靶序列特异性结合时,才能完成对目的基因的切割。PAM序列是CRISPR/Cas9系统发挥作用的先决条件。Cas9识别错误或其他模式低频PAM序列是产生脱靶的部分原因[19]。

图3 (A)1 bp插入(DNA凸起)(B)1 bp缺失(RNA凸起)示意图[18]
3.2 sgRNA
sgRNA由crRNA和tracrRNA组成,其中crRNA负责识别约20 bp的靶序列,tracrRNA能够指导crRNA与靶序列特异性结合。sgRNA的结构和长度会对脱靶效应或切割效率产生一定影响。在Cas9与PAM序列结合完成后,sgRNA与靶序列特异性结合,Cas9对靶序列进行切割。sgRNA是CRISPR/Cas9系统发挥作用的重要条件。由于CRISPR/Cas9系统是源于细菌等对噬菌体侵害的免疫机制,为保证将外源DNA全部清除,sgRNA本身便具有一定容错能力。研究表明,sgRNA自身可耐受1-5个碱基错配[20]。sgRNA与靶序列的错配是产生脱靶现象的最主要原因,前文所述的“DNA凸起”和“RNA凸起”一定程度上也可以认为是sgRNA与靶序列的错配。
3.3 其他
除以上所列举的影响脱靶效应的因素外,还存在一些其他干扰因素,如转染的细胞类型、染色质的结构、基因组背景影响等。研究发现,CRISPR/Cas9系统在斑马鱼胚胎细胞中的突变是高效的,通过质粒转染进行基因编辑,其靶向突变率可达86%[21]。但是在人K562细胞中,同样通过质粒转染,即使是在sgRNA出现错配的情况下,CRISPR/Cas9系统仍具有较高活性[16]。这表明CRISPR/Cas9系统存在着一定的细胞特异性。由于DNA在细胞中是以染色体的状态存在,靶序列以及PAM序列均被包埋在染色体内部,Cas9与染色体亲和特性也在一定程度上影响CRISPR/Cas9系统特异性[19]。同时,不同的基因组背景也会对系统的特异性产生一定影响,但具体机制还未被揭示。
4 降低脱靶效应的策略
影响脱靶效应的相关因素被成功揭示之后,一系列减少脱靶效应的新技术被成功创立。目前,减少脱靶效应的方法主要是优化或改进sgRNA、改造Cas9蛋白以及应用SpCas9蛋白类似物等。
4.1 sgRNA合理设计及修饰
sgRNA与靶序列的特异性结合是CRISPR/Cas9系统基因编辑的最关键因素。不合理的sgRNA设计会造成特异性降低、脱靶率升高。sgRNA设计应遵从以下原则:(1)最小化sgRNA与其他序列的相似性,且sgRNA与非靶序列存在超过3个错配;(2)至少2个错配位于非靶序列的种子区域内;(3)错配应是连续的或者间隔小于4 nt[22]。同时,实验表明sgRNA中GC含量≤35%时,sgRNA具有良好的特异性[23]。
在合理设计sgRNA的基础上,通过对sgRNA的修饰可进一步提高CRISPR/Cas9系统的特异性。Cho等[24]在sgRNA的5'端携带了两个额外的G,结果意外发现系统的特异性大幅提高。Hsu等[25]在靶向人EMX1和PVALB基因时,对tracrRNA尾部进行不同程度的延长,发现sgRNA延长后的基因插入水平比未修饰的sgRNA高5倍。这表明tracrRNA尾部的延长,在一定程度上增强了CRISPR/Cas9系统稳定性。此外,5'端截短1-3 bp的sgRNA仍然显示类似于全长sgRNA的切割活性[18]。同时,通过对sgRNA进行不同的化学修饰,例如嵌合sgRNA法,也能够达到降低脱靶效应的效果[26]。
4.2 Cas9突变体
Cas9是CRISPR/Cas9系统中的重要组成部分,最广泛使用的Cas9是源自化脓性链球菌的SpCas9。Cas9含有HNH及RuvC核酸酶结构域,使其中一个或将其全部失活,可分别获得D10A Cas9(Cas9 D10A mutation,Cas9n)[27]以及丧失核酸酶活性的Cas9(Deactivated Cas9,dCas9)[28]。成对的 Cas9n及dCas9可以代替Cas9,且成倍的提高系统特异性。Cas9蛋白剪切DNA过程,如图4。
Cas9与DNA的接触影响Cas9/sgRNA与DNA结合的稳定性以及对错配的耐受性。对SpCas9中负责与靶序列接触的关键氨基酸残基的改变,获得了高保真变体“SpCas9-HF1”[30]。改变 SpCas9与非靶链结合的氨基酸获得了增强型特异性SpCas9变体“eSpCas9”[31]。SpCas9-HF1、eSpCas9 均显著降低了脱靶效应,且保留了稳健的靶向切割。通过对REC3结构域的突变,Chen等[32]设计了一种新型超精确SpCas9变体“HypaCas9”。HypaCas9具有更优异的特异性,同时具有更高的靶向活性。此外,最近还获得了高保真度的“evoCas9”[33],扩展PAM兼容性的“xCas9”[34],可以同截短或延长的sgRNA组合使用的“Sniper-Cas9”[35]以及高特异的“Hifi Cas9”[36]。
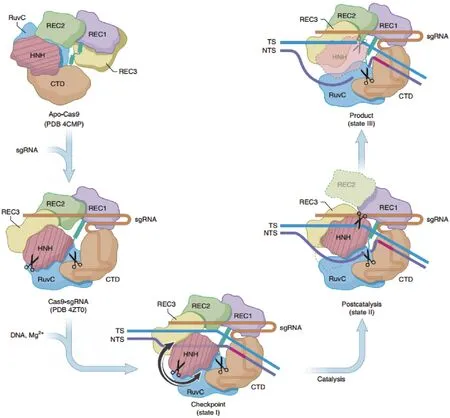
图4 Cas9蛋白剪切DNA全过程的结构变化[29]
4.3 Cas9类似物
除了Cas9突变体,SpCas9类似物也提供了一种降低脱靶效应的新途径。通过识别不同的、更复杂的PAM,SpCas9类似物获得了更高的特异性。SpCas9类似物来源广泛包括:金黄色葡萄球菌SaCas9[37]、 嗜 热 链 球 菌 St1Cas9[38]、St3Cas9[39]、脑膜炎奈瑟菌 NmCas9[40]、弗朗西斯菌 FnCas9[41]、空肠弯曲杆菌CjCas9[42]以及Cas12a家族中的毛螺旋菌 LbCas12a[43]和氨基酸球菌 AsCas12a[44]。此外,最近还发现BhCas12b能够表现出比SpCas9更高的特异性[45]。
虽然SpCas9类似物具有较高的特异性,但其较低的靶向性阻碍了其广泛应用。在众多类似物中,仅SaCas9、CjCas9存在与SpCas9相当的靶向性。SaCas9识别“NNGRRT”,可与20-24 nt sgRNA组合作用[46]。CjCas9识别“NNNNACAC”和“NNNNRYAC”,与GX22sgRNA共同作用时,显示了最佳靶向活性[42]。通过设计突变体可以改善SpCas9类似物较低的靶向性。目前,通过对SaCas9进行突变,获得的“SaCas9-KKH”可以提升靶向性2-4倍[47]。此外,还可以通过多种方法扩展SpCas9类似物的应用,如通过抗CRISPR蛋白与NmCas9的结合,可实现对NmCas9活性的调节[48];通过dCas9与FnCas9共同作用,可提升FnCas9的靶向活性等[49]。
4.4 Cas9/sgRNA浓度改变
酶的特异性和活性强度通常高度依赖于反应条件,高酶浓度下会弱化酶的特异性。Hsu等[25]在靶向人EMX1基因的实验中,减少转染细胞的质粒数(编码Cas9蛋白和sgRNA),结果发现系统的特异性大大增强。这表明通过改变Cas9/sgRNA复合物的浓度实现最佳特异性的可行性。然而Cas9/sgRNA浓度的降低会导致CRISPR/Cas9系统切割效率的降低,所以需要通过实验确定最佳Cas9/sgRNA浓度。在使用质粒转染细胞时,通过减少细胞中sgRNA的数量,可直接限制细胞内Cas9/sgRNA复合物浓度。此外,在使用质粒转染细胞时,采用不同启动子影响sgRNA转录,也能够降低细胞内Cas9/sgRNA浓度,减少脱靶[50]。
5 脱靶检测技术
脱靶检测技术是一系列针对CRISPR/Cas9系统作用机制研发的用于确定CRISPR/Cas9系统基因编辑准确性的检测工具。脱靶检测在揭示CRISPR/Cas9系统的脱靶机制以及进一步提高系统靶向性的研究中具有重要作用。
早期的脱靶检测技术是由软件预测和测序组成,如Sanger测序、NGS测序、全外显子组测序等[51-52]。该类技术的原理是针对预测获知的脱靶位点进行测序,以确定是否在这些位点发生了非特异性结合。Sanger测序法是检测CRISPR/Cas9系统脱靶的常用方法之一。首先,通过Cas-OFFinder等[53]脱靶预测软件进行预测,获得可能的脱靶位点。然后,对预测的脱靶位点进行PCR扩增、测序,从而确定该位点是否发生脱靶突变。该类技术存在明显的偏向性,其主要针对的是软件预测的脱靶位点,而软件预测往往容易造成部分脱靶位点的遗漏。
由易错的NHEJ修复Cas9诱导的DSB时,往往会发生脱靶突变。检测Cas9脱靶的最直接方法是跟踪基因组中的DSB。通过对DSB的标记实现了全基因组无偏脱靶检测,如IDLVs、BLESS、GUIDE-seq技术等[54-56]。这类技术的原理是通过将特定的双链DNA或生物素接头整合到DSB中,从而达到检测目的。GUIDE-seq是无偏检测脱靶效应的方法之一。首先,需要将特定的双链寡核苷酸(Double stranded oligodeoxynucleotides,dsODN)整合到断裂位点。然后提取基因组DNA,随机打断;对含有dsODN片段的序列进行扩增、富集。最后,测序后分析切割位点,评估脱靶效应[57]。该类技术虽然实现了全基因组无偏检测,但是其只能检测断裂时期的DSB,对于已经修复或者未发生的DSB则不能检测。
近年来,利用Cas9蛋白能够在体外消化DNA的特性,使用发夹接头或生物素等标记DNA片段,开 发 了 Digenome-seq、Circle-seq、SITE-seq等 技术[58-60]。该类技术的原理是利用Cas9体外核酸酶特性,在体外对基因组DNA进行切割,产物经处理后,通过测序或其他手段,实现对脱靶位点的筛选。Digenome-seq是利用Cas9体外消化DNA特性检测脱靶效应的技术之一。Cas9体外消化基因组DNA、测序;比对具有相同末端的序列,通过软件分析即可评估脱靶效率[61]。该类技术同样从全基因组角度实现了无偏检测,且精度较前两类技术更高,唯一存在的问题是Cas9在体内和体外发挥作用时可能会存在一定的差异。
此外,脱靶检测方法还有利用T7EⅠ酶、Surveyor酶等对错配碱基切割的酶切法;利用dCas9与靶序列和非靶序列结合,结合测序手段的Chipseq技术以及基于染色体易位原理的HTGTS检测法等。最近,针对各类脱靶检测方法存在的问题,开发了一种普遍适用的无偏脱靶识别方法DISCOVERSeq。DISCOVER-Seq的优势在于利用了DNA修复蛋白MRN复合物的亚基MRE11,MRE11与DNA的结合在插入缺失之前就可达到了峰值,结合Chipseq与定制软件BLENDER,通过软件得分便可实现对脱靶事件的高度特异性鉴定[62]。DISCOVER-Seq提供了一种定义和量化整个生物体中基因编辑脱靶效应的一般策略,从而为促进体内基因编辑疗法的开发提供了蓝图。
6 总结与展望
作为主流基因编辑工具,CRISPR/Cas9的应用大大加快了分子生物学、植物学、生命科学等研究工作。在生物学中,CRISPR/Cas9用于转录调节、基因成像以及表观遗传学研究,这有助于进一步理解真核基因表达;在植物学中,CRISPR/Cas9加速了作物品种改良工作,缩短了作物育种年限,极大缓解了全球粮食危机;在医学中,CRISPR/Cas9显示出治愈困扰人类许久的疑难杂症的极大潜力,已被广泛用于细胞及动物模型的创建、药物设计筛选等,这是CRISPR/Cas9迈向医学领域的第一步。目前影响脱靶效应因素的研究集中在sgRNA与靶序列的结合以及Cas9对PAM序列的识别,其他诸如盐离子浓度等对脱靶结合的影响还未被揭示。通过优化/改进sgRNA、突变Cas9和采用Cas9类似物,已成功减少非特异性结合的发生,但脱靶率的降低有时也会导致靶向切割效率的降低。如何在进一步降低脱靶效应的同时保持原有切割效率将成为新的研究重心。此外,伴随精准测序技术的实现,更为精准的检测技术也必将不断推陈出新。随着脱靶影响因素、降低策略及脱靶检测技术研究的不断深入,未来CRISPR/Cas9系统必将会应用在更广泛的领域造福人类。
