采用ForenSeqTM DNA Signature Prep试剂盒进行祖先推断
2019-12-27
(中山大学中山医学院法医学系,广东 广州 510089)
祖先信息标记(ancestry informative marker,AIM)是人类基因组中在族群之间显示出分布模式差异的遗传标记,其等位基因频率与基因型频率在不同地域或民族群体间差异明显,因而可用作祖先信息推断。单核苷酸多态性(single nucleotide polymorphism,SNP)[1]、插入/缺失(insertion/deletion,InDel)多态性[2]、Y染色体短串联重复(Y chromosomal short tandem repeat,Y-STR)[3]、线粒体 DNA(mitochondrial DNA,mtDNA)[4]、微单倍型(microhaplotype)[5]等均可作为AIM,其中SNP因为在人类基因组中数量丰富且有国际人类基因组单体型图计划、千人基因组计划等相应的数据库支持,成为近年来筛选AIM的主要对象,这类SNP被称为祖先信息SNP(ancestry informative SNP,aiSNP)[6]。在法医学应用中,对aiSNP分析可以在重大灾害事件或案件调查中缩小调查范围,加快失踪人员或灾害受害者的识别进程,在缺乏目击证人证词或数据库无法比对时提供调查线索,因此被法医学界关注和重视[7-8]。目前已陆续报道了一些基于毛细管电泳(capillary electrophoresis,CE)平台的aiSNP分型体系用于预测个体祖先和进行族群推断[7,9]。
二代测序(second generation sequencing,SGS)技术革新了传统的Sanger测序方法,其基本原理是边合成边测序,能够同步对多样本、多遗传标记进行较高覆盖度的分析,近年来在法医学领域的应用逐渐增多[10-11]。其中,MiSeq FGx法医基因组学系统(美国Illumina公司)是目前在法医学领域常用的二代测序技术平台之一,基于此平台的ForenSeqTMDNA Signature Prep试剂盒系统能够同时扩增231个法医学检验相关的DNA遗传标记,包括58个STR、Amelogenin、94个个体识别 SNP(individual identification SNP,iiSNP)、56个aiSNP和24个表型信息SNP(phenotype informative SNP,piSNP)(aiSNP和piSNP有两个位点重叠)[12-13]。
我国是一个多民族国家,根据第六次全国人口普查数据显示,除了汉族人口占多数以外,蒙古族、藏族、维吾尔族3个少数民族的人口均超过500万,有比较集中的居住地,语言、服饰和习俗标识明显[14]。此外,作为对外开放程度高、经济较发达的城市,广州近年居住有数十万非洲人,其中尼日利亚裔约占1/3[15]。因此,本研究基于MiSeq FGx法医基因组学系统二代测序平台,采用ForenSeqTMDNA Signature Prep试剂盒的aiSNP标记,以尼日利亚群体和中国的4个群体(汉族、蒙古族、藏族和维吾尔族)作为研究对象,评估其推断祖先来源的效果。
1 材料与方法
1.1 样本收集
根据知情同意原则采集志愿者外周血,置于滤纸片或FTA卡上制备为血痕。5个群体共计85个样本,其中包括:河北汉族个体23个,编号为H1~H23;内蒙古自治区蒙古族个体13个,编号为M1~M13;西藏自治区藏族个体12个,编号为Z1~Z12;新疆维吾尔自治区维吾尔族个体15个,编号为W1~W15;尼日利亚个体22个,编号为N1~N22。本研究经中山大学中山医学院伦理委员会批准。
1.2 文库构建
使用1.2 mm直径打孔器取血痕2片,使用ForenSeqTMDNA Signature Prep试剂盒(美国Illumina公司)的B系列引物(DNA Primer Mix B,DPMB)进行文库构建。PCR组分包括DPMB 5.0μL,PCR反应混合液1 4.7 μL,酶混合物0.3 μL。热循环条件为:98℃变性3 min;96℃ 45 s,54℃ 2 min,68℃ 2 min,共8个循环;68℃末次延伸3min,10℃保存。对以上扩增产物添加样本特异标签(i5和i7)并实现靶点富集,热循环参数为:98℃ 30 s;98℃ 20 s,66℃ 30 s,68℃ 90s,共循环15次;68℃ 10min,10℃保存。利用样品纯化微珠(sample purification beads,SPB)和标准化文库珠(library normalization beads,LNB)对文库进行纯化和标准化。最后将每个样品的文库5μL混合到一个微量离心管中。
1.3 DNA测序
将7 μL混合后的文库、951 μL杂交缓冲液(hybridization buffer,HT1)、2μL稀释后的人类测序对照(human sequencing control,HSC)混合,使用 MiSeq FGxTMReagent试剂盒在MiSeq FGxTM测序仪上进行测序。每批检测均设置2800M为阳性对照DNA(美国Promega公司),无菌水为阴性对照。
1.4 数据分析
测序原始数据自动导入ForenSeqTM通用分析软件(universal analysis software,UAS)v1.2.16173(美国Illumina公司)进行分析,按照默认分析阈值和解释阈值进行56个aiSNP位点的等位基因判读。选择UAS的“Phenotype Estimation”分析页面进行个体表型和生物地理祖先推断。UAS祖先分析模块主要利用千人基因组计划的四大群体(欧洲、东亚、非洲和混合美洲)作为训练样本集,包括欧洲个体321个、东亚个体251个、非洲个体209个、混合美洲个体156个。根据56个aiSNP基因型数据与每个待检样本进行主成分分析(principle component analysis,PCA)。此外,本研究以三个非混合人群(欧洲、东亚和非洲人群)作为参考群体,基于56个aiSNP等位基因频率数据(http://spsmart.cesga.es/),对本研究中每个个体分别计算在三个参考群体的群体匹配概率(assignment match probability,AMP)和似然比(likelihood ratio,LR),根据最大群体匹配概率和似然比确定其祖先分配区域[16-17],按照江丽等[16]设定的判断方法:LR>100时,AMP值排第一位的人群为未知个体的来源人群;当LR≤100时,AMP值的前两位人群均不排除。参考千人基因组数据库(http://www.internationalgenome.org/)中欧洲、东亚和非洲人群的56个aiSNPs的分型,采用PAST v3.10进行PCA[18]。利用Structure 2.3.4软件(http://pritchardlab.stanford.edu/structure.html)分析本研究5个群体的祖先组成成分,设置0.70作为归属到某种人群的标准。设置运行长度为10 000,迭代次数为10 000,选择相关等位基因频率模型[19],通过Structure Harvester(http://taylor0.biology.ucla.edu/struct_harvest/)确定最佳K值[20],采用 Distruct 1.1(https://web.stanford.edu/group/rosenberglab/distruct.html)生成最终图形[21]。
2 结 果
2.1 测序质量
本次研究按照群体类别共进行了5批测序,每批实验的2800M阳性对照DNA的分型均与试剂盒标准分型结果一致,显示该体系的准确性和稳定性。85个样本的测序深度(depth)见图1A,从图中可以看出最低的两个位点是rs310644和rs3811801,但都大于UAS中默认的分析阈值(10×)和解释阈值(30×),所有位点的平均测序深度为566×。图1B为杂合子等位基因覆盖比(allele coverage ratio,ACR),从该图中可以看出除位点rs1426654低于默认阈值(0.5)外,其他位点均较高,均值为0.86,整体测序质量良好。
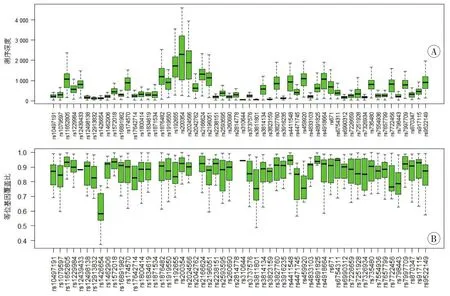
图1 本研究85个样本56个aiSNP的测序质量
2.2 ForenSeqTMUAS分析
经MiSeq FGx法医基因组学系统配套的UAS生物地理祖先分析模块,获得每个样本基于非洲、东亚、欧洲、混合美洲4个参考人群的PCA位置图以及每个样本与最近的群体聚集中心(centroid)的距离、千人基因组中与待测样本距离最近的群体样本列表、离检测样本最近的聚集中心的参考样本列表。85个样本通过UAS分析的PCA叠加图见图2,可以看到:河北汉族、西藏自治区藏族个体通常落入东亚群体内,也有个体处于东亚和混合美洲群体的边缘;内蒙古自治区蒙古族个体主要聚集在东亚群体内,但也有少数样本处于东亚和混合美洲群体之间;新疆维吾尔自治区维吾尔族则主要聚集到东亚群体和混合美洲群体之间的位置;而尼日利亚个体则全部聚集在非洲群体内。
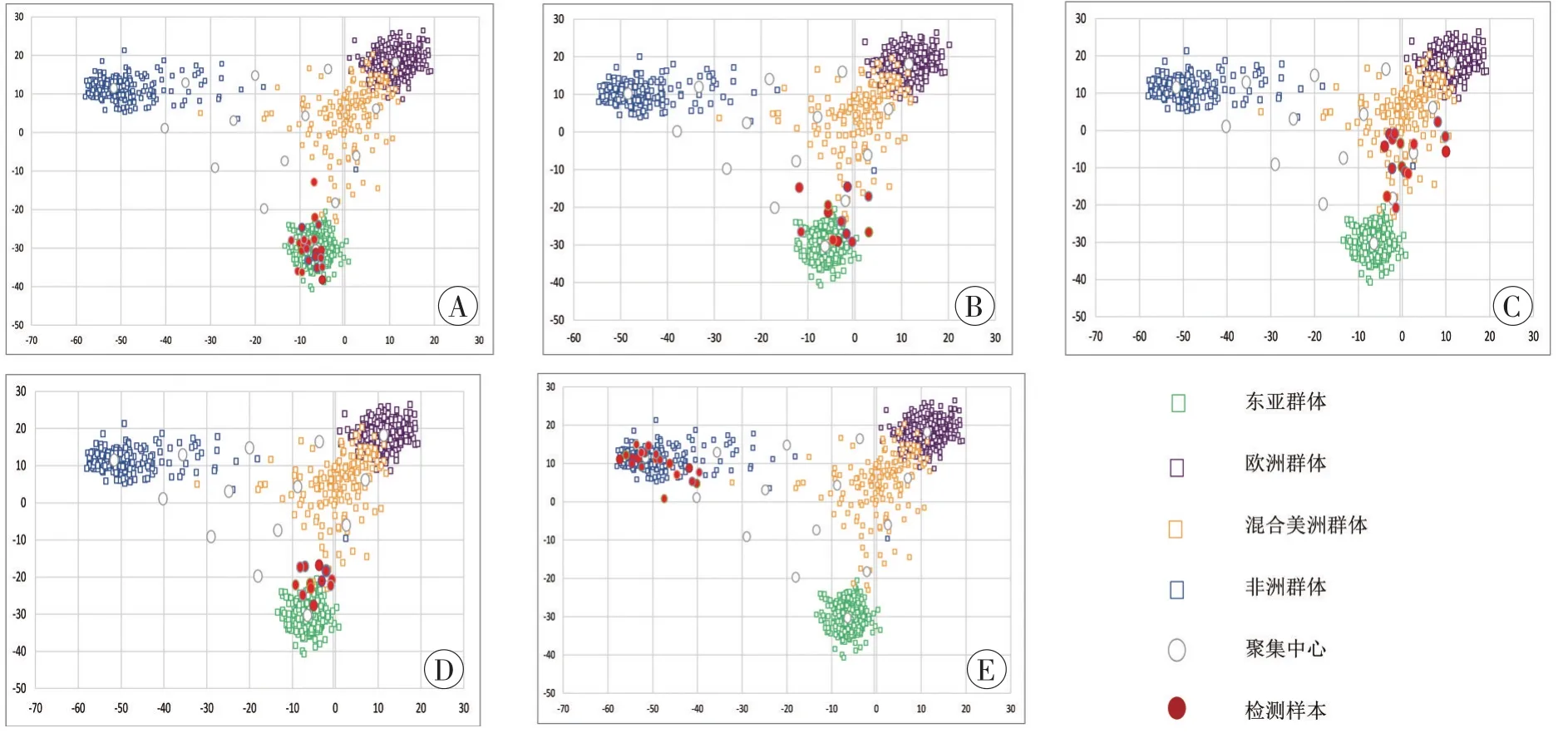
图2 UAS分析获得的本研究5个群体样本与祖先预测PCA叠加图
2.3 PCA
如图3所示,主成分1(PC1)、主成分2(PC2)分别占总变化的49.53%和7.38%。基于前两个因素,河北汉族、西藏自治区藏族和内蒙古自治区蒙古族聚为一簇,新疆维吾尔自治区维吾尔族单独为一簇,并且4个群体都处于同一侧,而尼日利亚群体处于另外一侧,与中国的4个群体相距较远。该分析结果与UAS祖先分析结果一致。
2.4 AMP和LR
以欧洲、东亚和非洲三个非混合种群作为参考群体,根据最大群体匹配概率和似然比确定本研究每个个体的祖先分配区域,结果如表1所示,河北汉族、西藏自治区藏族、内蒙古自治区蒙古族中的所有个体均被分配到东亚种群(100%),来自尼日利亚的群体被分配到非洲种群(100%)。新疆维吾尔自治区维吾尔族与其他4个研究群体的祖先地理分配比例有明显的差异,主要预测来源于东亚和欧洲群体,其分配比例分别为60%和40%。
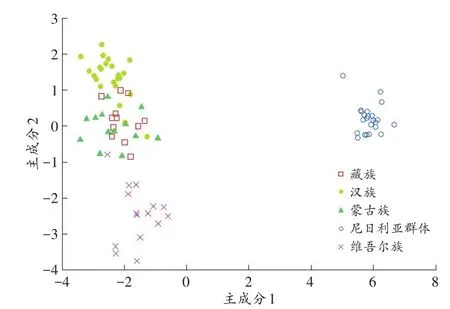
图3 本研究5个群体样本的PCA图
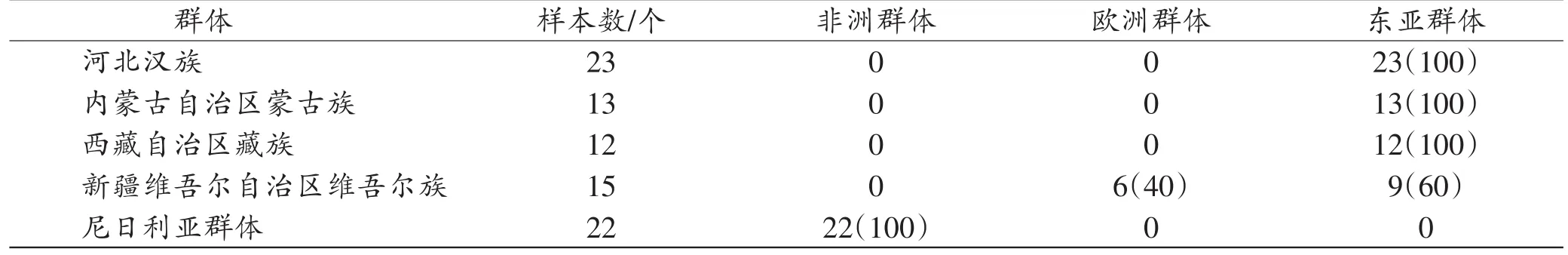
表1 根据AMP和LR推断生物地理祖先 [n(%)]
2.5 祖先成分分析
由图4A可见,在本研究中获得最佳K值为3的Structure分析图,欧洲、东亚和非洲群体可以显现和区分。图4B显示,本研究5个群体最佳K值为3,主要分为三个聚类,其中河北汉族、新疆维吾尔自治区维吾尔族和尼日利亚群体各自的祖先成分相对比较集中,内蒙古自治区蒙古族和西藏自治区藏族的少数个体含有极少比例的混合成分。结合表2可见:本研究中河北汉族、西藏自治区藏族群体的东亚成分比例分别为0.987和0.960;内蒙古自治区蒙古族主要含有东亚成分(88.4%),含有较少比例的欧洲成分(10.0%);尼日利亚群体的非洲群体成分比例为99.7%;而新疆维吾尔自治区维吾尔族则明显表现出东亚和欧洲的混合成分,其比例分别为53.3%和45.7%。
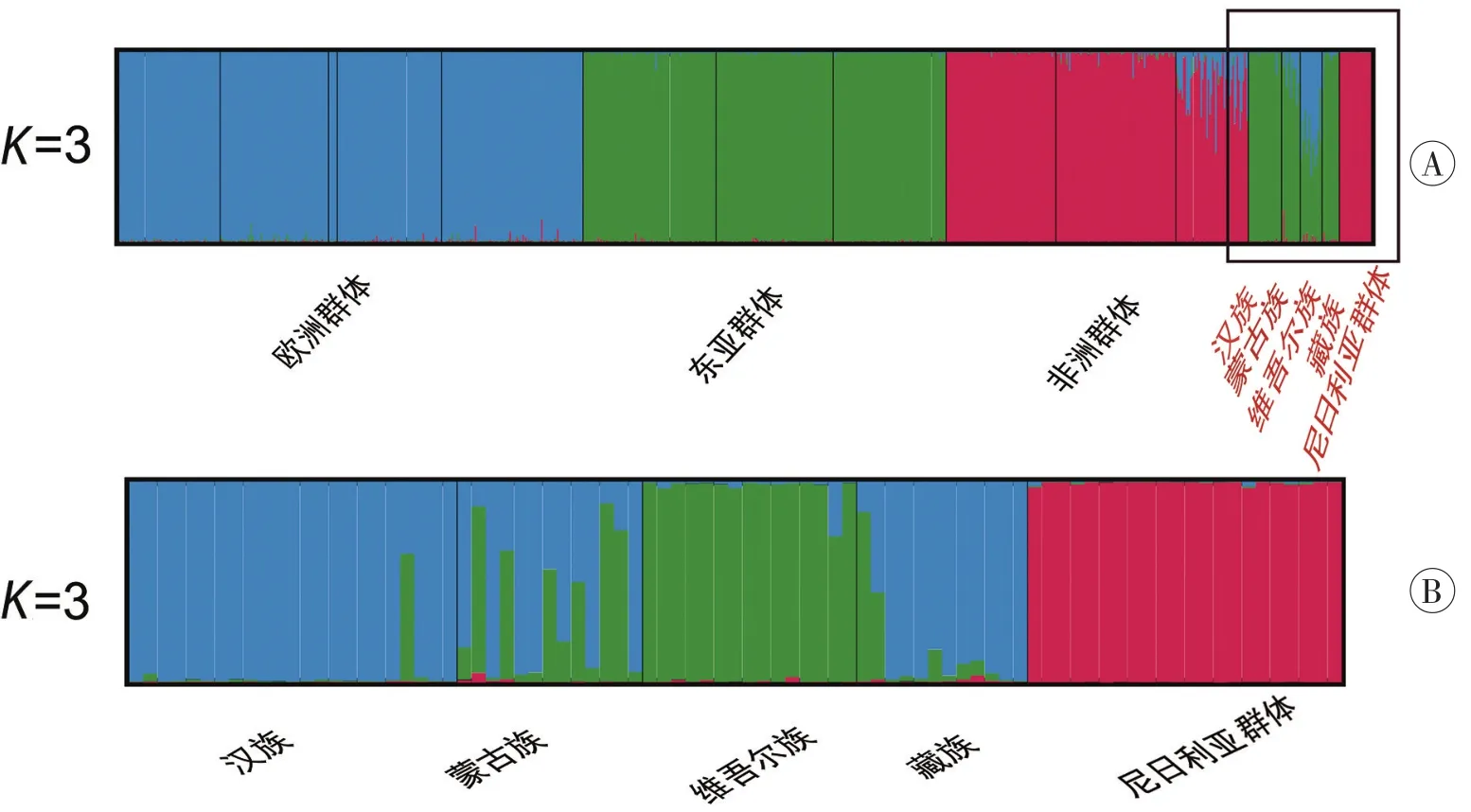
图4 本研究的5个群体与3个参考群体56个aiSNP分型的Structure分析图
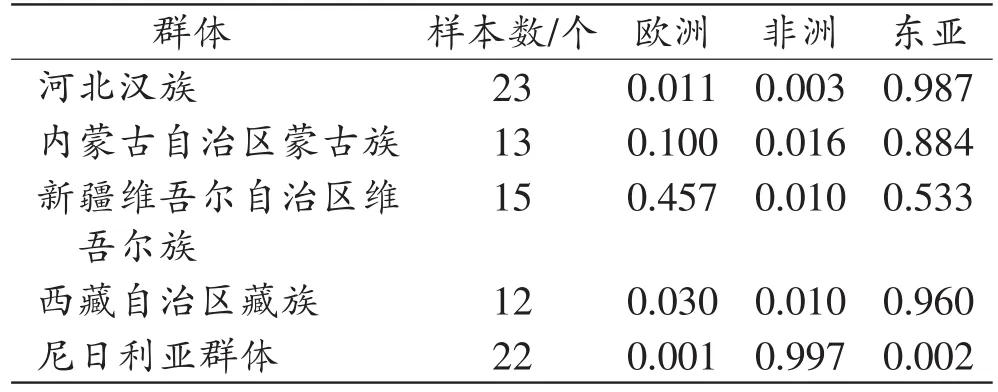
表2 本研究的5个群体的祖先成分比例
表3显示了本研究每个个体的祖先成分分析结果,设定0.70作为判定混合人群的标准,结果显示尼日利亚个体中非洲群体成分全部大于0.980,100%的个体显示出非洲群体来源(22/22);河北汉族、内蒙古自治区蒙古族、西藏自治区藏族个体中东亚人群成分全部大于0.700,100%被归入东亚群体来源。而新疆维吾尔自治区维吾尔族个体中仅有2个样本(W4和W14)的东亚成分比例大于0.700,即只有13.3%(2/15)的样本被归入东亚群体来源,其余样本同时具有相对较高比例的欧洲来源(0.294~0.649),显示出明显的混合遗传的特点。
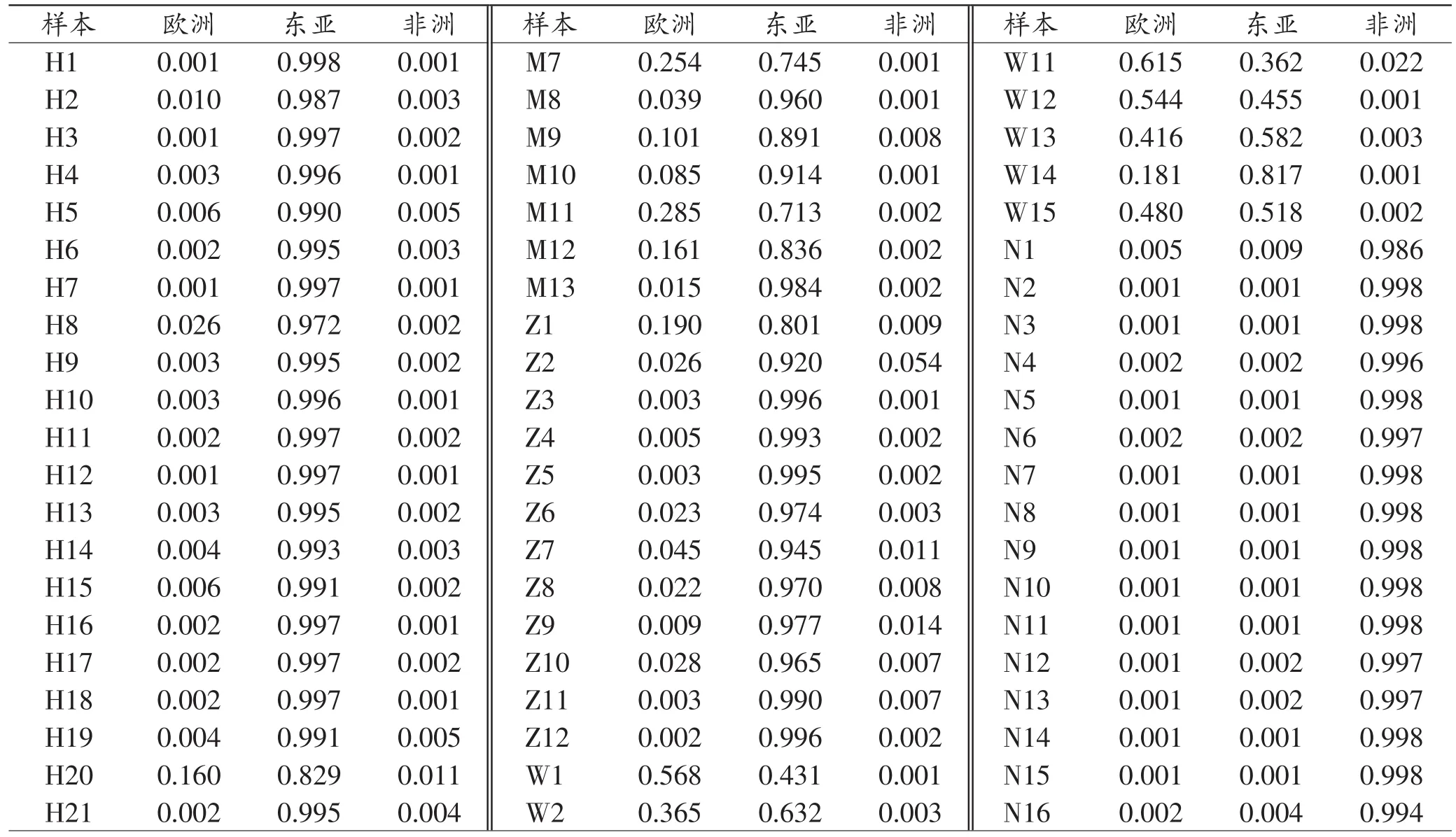
表3 本研究85个样本的祖先成分分析
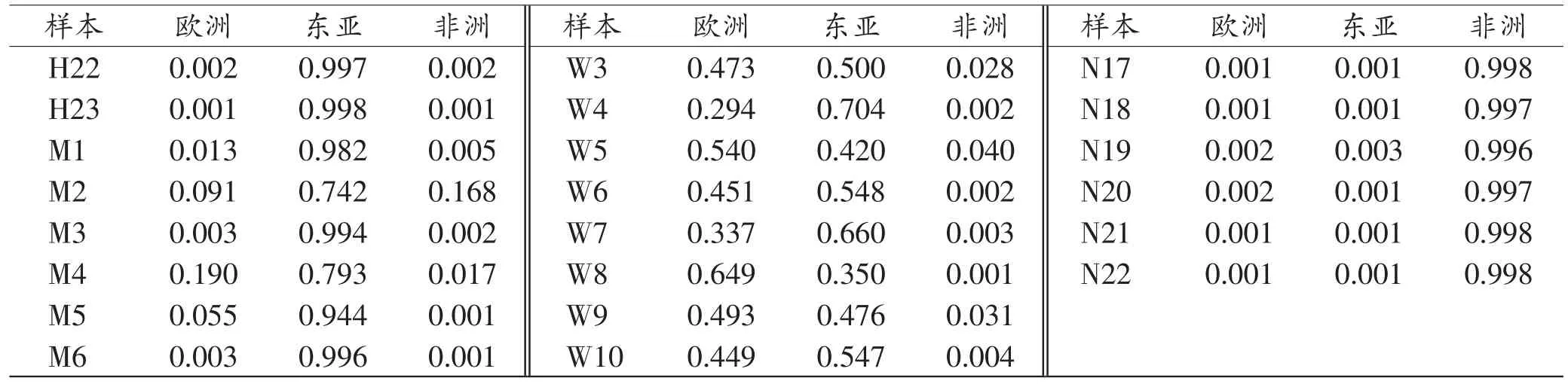
续表3
3 讨 论
本研究用于祖先推断的56个aiSNP位点最早由KIDD等[17]所报道,被美国Illumina公司整合入ForenSeqTMDNA Signature Prep试剂盒。基于二代测序平台能在检测STR、SNP分型进行个体识别时同步进行族群推断,提高了单一检测体系获得的遗传信息。
本研究基于MiSeq FGx法医基因组学系统检测平台,对中国河北汉族、内蒙古自治区蒙古族、西藏自治区藏族和新疆维吾尔自治区维吾尔族以及居住在广州的尼日利亚个体进行56个aiSNP位点测序和基因分型,并采用了该体系配套的UAS及其他公共软件通过PCA、AMP、LR计算,Structure分析对各个样本的生物地理祖先进行推断。其中PCA分析显示:河北汉族和西藏自治区藏族人群与东亚参考群体聚集在一起;尼日利亚人群与非洲参考群体聚集在一起;内蒙古自治区蒙古族主要聚集在东亚参考群体,少数个体位于东亚和混合美洲参考种群之间;新疆维吾尔自治区维吾尔族人群则与混合美洲聚集在一起并处于东亚和欧洲群体之间。在UAS软件中混合美洲群体作为一种特殊的混合人群,主要包括洛杉矶的墨西哥人、波多黎各的波多黎各人、麦德林的哥伦比亚人和秘鲁利马的秘鲁人等混合群体。有文献[19]报道,该混合种群的祖先成分包括东亚和欧洲的混合组成,通过Structure分析具有与南亚和中亚相似的遗传成分[22]。为了获得较明确的预测,本研究选择东亚、欧洲、非洲3个非混合人群作为洲际参考群体,根据群体匹配概率和似然比推断祖先来源的结果,显示河北汉族、内蒙古自治区蒙古族、西藏自治区藏族个体100%被归入东亚祖先来源,100%的尼日利亚个体被归入非洲来源。通过Structure分析获得每个样本的祖先成分比例,若参考RAMANI等[23]的报道,设定0.75作为判定归属到某种人群的标准,河北汉族、西藏自治区藏族个体也将100%被归入东亚祖先来源,但内蒙古自治区蒙古族中只有76.9%(10/13)的个体被归入东亚人群,与通过AMP和LR的预测结果存在差异。结合本研究UAS和LR法的分析结果,尝试以0.70作为标准,则可将内蒙古自治区蒙古族个体100%划分到东亚来源。而在上述几种方法中,新疆维吾尔自治区维吾尔族均明显地表现出东亚和欧洲的混合来源特点,与已有文献报道[22,24]新疆维吾尔自治区维吾尔族祖先组成成分是东亚和欧洲的混合相一致。文献[24]指出,预测祖先来源分析时,特别是对于混合群体的祖先来源推断及祖先成分分析,如果只考虑似然比的值,对地理位置相近的种群,由于持续的人口迁移和通婚就会导致分类错误。在这种情况下,Structure分析祖先成分可以较明确地体现其混合来源构成,更客观地判断个体的祖先起源。因此,在进行祖先来源预测中可结合两种方法进行分析。
本研究采用上述方法对5个群体的分析结果一致,即:尼日利亚群体与中国的4个民族群体之间具有明显的遗传差异,通过该检测体系全部归属到非洲种群;河北汉族、西藏自治区藏族和内蒙古自治区蒙古族的祖先预测显示主要来源于东亚;新疆维吾尔自治区维吾尔族预测为东亚和欧洲的混合成分。通过该体系可将新疆维吾尔自治区维吾尔族与我国其他3个民族(河北汉族、西藏自治区藏族、内蒙古自治区蒙古族)进行区分,但尚不能更进一步具体地明确河北汉族、西藏自治区藏族、内蒙古自治区蒙古族之间的遗传差异。
综上所述,ForenSeqTMDNA Signature Prep试剂盒中的aiSNP体系从洲际群体水平对本研究的5个群体之间的遗传关系和祖先预测的效能是可靠的,但尚不能区分种群中的亚群体之间的遗传关系和祖先来源,需要开发验证更多的参考种群数据和更高差异的东亚特定aiSNP标记,以便在法医祖先推断中提供更高的族群分辨率,进行更精细的祖先推断[22]。
