巴哈雀稗幼苗叶片转录组分析
2019-12-09张宇君尚以顺王普昶
张宇君 尚以顺 王普昶


摘 要:利用Illumina HiSeq 2000高通量测序技术对巴哈雀稗叶片进行转录组测序,共计获得42 844 132个序列读取片段,包含碱基序列信息6 426 619 800个。对读取片段进行序列组装,得到99 235个单基因簇。围绕GC含量、测序质量、长度分布的单基因簇评估测序数据均显示质量好、可信度较高。与Nr数据进行序列同源性比对结果显示,有53 886个单基因簇与其他植物的已知基因具有不同程度的同源性。结合GO数据库对巴哈雀稗的单基因簇进行功能分类,大致可分成为细胞组分、分子功能和生物学过程3大类共计70个分支,其中大量的单基因簇主要与细胞进程、结合活性及细胞成分有关。将单基因簇与COG数据库进行比对分析,依据其功能可大致分为25类。以KEGG数据库为参考,依据代谢途径将单基因簇定位到31个代谢途径分支上,具体包含核糖体代谢途径、碳代谢途径、氨基酸生物合成途径、内质网蛋白加工、剪接体和甜菜红色素生物合成等。通过SSR位点查找在巴哈雀稗99 235个单基因簇中共找到12 062个SSR位点。其SSR不同重复基序类型中,A/T出现频率最高,其次是AT/GA和AG/CT。
关键词:巴哈雀稗;转录组;基因注释;SSR
中图分类号:Q789文献标识码:A文章编号:1006-060X(2019)10-0001-07
Abstract: The transcriptome of Paspalum notatum leaves was sequenced using Illumina HiSeq 2000 high-throughput sequencing technology, 42 844 132 sequences were readed, containing 6 426 619 800 bases. Sequence assembly of the read fragments yielded 99 235 unigenes. The evaluation of unigene based on GC content, sequencing quality and length distribution showed that the data volume and quality of sequencing were good and the reliability was high. Sequence homology analysis compared with Nr data revealed that 53 886 unigenes had different degrees of homology with known genes of other plants. Unigenes of Paspalum notatum were roughly divided into 70 branches of cell components, molecular functions and biological processes by function classification combined with the GO database, a large number of unigenes mainly related to cell processes, binding activities and cellular components. After comparing and analyzing the unigenes with COG databases, they were roughly classified into 25 categories according to their functions. Based on the KEGG database, unigenes were mapped to 31 metabolic pathway branches including ribosomal metabolism, carbon metabolism, amino acid biosynthesis, endoplasmic reticulum protein processing, splice and beet red color biosynthesis. A total of 12 062 SSR loci were found in the 235 unigenes of the Paspalum notatum by SSR locus analysis. A/T had the highest occurrence frequency among the different repeat motif types of SSR followed by AT/GA and AG/CT. This study provided an important source of genomic data for the molecular biology research of the genus Paspalum notatum.
Key words: Paspalum notatum; transcriptome; gene annotation; SSR
巴哈雀稗(Paspalum notatum)是一种原产于南美洲的禾本科雀稗属黍族多年生草本植物。于20世纪50年代初首次引入台湾,适宜在年降水量高于750 mm的热带和亚热带地区生长,尤其在中国的云、贵、川、湘等南方地区种植较多。目前已在中国西南、华南近10个省区推广,并被成功应用于三峡库区的水土保持和生态防护工程中[1]。巴哈雀稗的抗逆性强,具有耐贫瘠、抗旱、固地性强等特点,在治理水土流失中发挥着重要作用[2]。因其对土壤的要求低,在肥力缺乏的沙质土壤中仍能顽强生长,常被选作公路、堤坝、机场跑道绿化的优选草种,具有较大的应用开发潜力。目前关于巴哈雀稗的研究主要集中在水土流失治理、荒山荒坡綠化、果园覆盖、饲草料等方面[3],而有关巴哈雀稗遗传多样性、分子标记开发等分子方面的研究相对较少。开展巴哈雀稗的转录组学研究,能为牧草、草坪草分子领域的相关研究提供基础数据。
近年来,在植物细胞代谢过程和生命活动规律研究中被广泛应用的转录组测序技术来源于以大量基因序列或分子数据库信息为前提的传统芯片杂交平台[4],该平台的特点在于:不仅能够准确、快速、全面地获得选定植物在特定状态下某一组织的全部转录组信息[5],同时还能挖掘某些重要的功能基因。研究以采自贵州省罗甸县龙坪镇的野生巴哈雀稗为试验材料,利用高通量测序技术获取巴哈雀稗幼苗葉片的转录组序列,以期通过序列组装、功能注释和深度分析,为今后开展有关巴哈雀稗的分子生物学研究提供素材。
1 材料与方法
1.1 试验材料
供试野生巴哈雀稗采自贵州省罗甸县龙坪镇(106°41′56.99″E,25°23′8.93″N),由贵州省农业科学院草业研究所提供。
1.2 试验方法
1.2.1 RNA提取与检验 随机选取10株使用Hoagland培养液培养至3叶一心时期的巴哈雀稗幼苗叶片,充分混合研磨后采取TRIzol法提取叶片的总RNA。采用NanoDrop-2000浓度测定仪对总RNA样品进行检测评估,以确保样品质量符合要求。
1.2.2 cDNA文库构建和测序RNA 样品经检验合格后,送上海生工生物工程有限公司进行cDNA文库构建和Illumina HiSeq 2000测序。
1.2.3 转录组组装 将获得的原始序列采用Trimmomatic
进行过滤,得到Clean数据,使用Trinity将Clean数据de novo组拼接组装成转录本,再对Trinity拼装得到的转录本去冗余,取每个转录本聚类中最长序列作为单基因簇。
1.2.4 基因功能注释 使用BLAST程序将获得的单基因簇与NR数据库进行序列相似性比对,然后选取最佳的功能注释;利用WEGO软件对所有的单基因簇进行GO功能分类统计;对巴哈雀稗单基因簇分别进行COG功能分类和KEGG代谢途径分析。
1.2.5 简单重复序列 对筛选获得的大于1 kb的单基因簇序列利用MISA软件进行SSR分析。
2 结果与分析
2.1 巴哈雀稗转录组的序列组装与分析
巴哈雀稗幼苗叶片转录组测序共计获得6 426 619 800 bp的序列信息,其中包含序列读取片段42 844 132个,每一片段的长度为150 bp;GC含量的平均值为62.62%;碱基Q30为92.18%。这表明该转录组测序数据量质量较好。
对巴哈雀稗的reads片段进行聚类组装,获得177 162个转录本,最长的为10 795 bp,最短的有201 bp,平均长度为771.38 bp,N50为1 186 bp(表1)。其中,有50 320个转录本长度在200~300 bp的范围内,占比28.40%;300~500 bp的转录本有36 902个,占比20.83%;500~1 000 bp的转录本有43 377个,占比24.49%;1 000~2 000 bp的转录本有35 280个,占比19.91%;≥2 000 bp的转录本有11 283个,占比6.37%(表2)。
在转录本数据的基础上,进一步对序列进行组装,又获得99 235个单基因簇,最长的有10 795 bp,最短的为201 bp,平均长度为641.96 bp(表1)。其中,单基因簇长度在200~300 bp的有39 470个,占比39.77%;单基因簇长度在300~500 bp的有22 352个,占比22.52%;长度在500~1 000 bp的单基因簇有18 822个,占比18.97%;1 000~2 000 bp的单基因簇有13 556个,占比13.66%;≥2 000 bp的单基因簇有5 035个,占比5.07%(表2)。
2.2 巴哈雀稗的单基因簇分析
2.2.1 单基因簇序列相似性分析 将组装获得的巴哈雀稗单基因簇与Nr数据库利用BLAST程序进行序列相似性比对。结果发现,有53 886个单基因簇均可在Nr数据库中找到相似序列;其中,巴哈雀稗与谷子(Setaria italica)的相似序列在匹配的近缘物种中所占比例最高,有14 130个,占比26.22%;高粱(Sorghum bicolor)有10 594个,占比19.66%;玉米(Zea mays)有6 908个,占比12.82%;二穗短柄草(Brachypodium distachyon)有3 569个,占比6.62%;大麦(Hordeum vulgare)有3 400个,占比6.31%;节节麦(Aegilops tauschii)有2 366个,占比4.39%;水稻(Oryza sativa)有1 913个,占比3.55%(图1)。
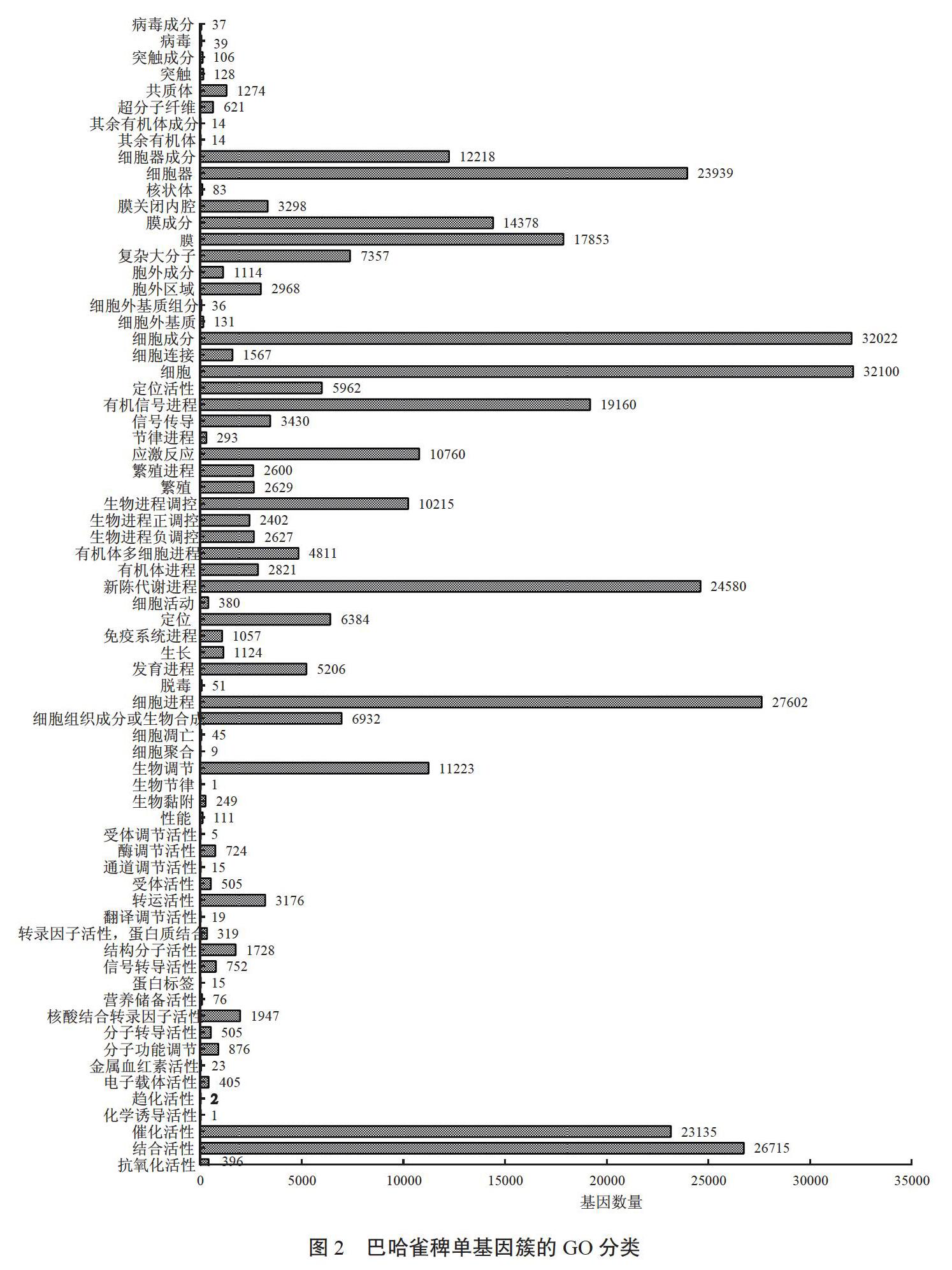
2.2.2 单基因簇的GO分类 结合GO数据库与NCBI
数据库的功能注释信息,对巴哈雀稗的单基因簇进行功能分类和表达基因功能分布特征分析,结果如图2所示,巴哈雀稗单基因簇具体可以划分为70个不同的功能组,归属于3大类:(1)生物学过程,有152 664个单基因簇;(2)细胞组分,有151 297个单基因簇;(3)分子功能,有61 339个单基因簇。其中,细胞(32 100个)、细胞成分(32 022个)、细胞进程(27 602个)、结合活性(26 715个)、新陈代谢进程(24 580个)、细胞器(23 939个)和催化活性(23 135个)功能组中涉及的单基因簇较多;而细胞聚合(9个)、受体调节活性(5个)、趋化活性(2个)、化学诱导活性(1个)和生物节律性(1个)功能组中所涉及的单基因簇较少。
2.2.3 单基因簇的COG分类 对巴哈雀稗单基因簇进行COG分类和功能预测,结果如图3所示,具体可分为25类;其中,细胞运动类单基因簇数量最少,仅有9个;核结构类和胞外结构类的单基因簇较少,分别只有103和77个;一般功能预测类和“翻译、核糖体结构和生物发生”类的单基因簇稍多,分别有2 924和2 053个;翻译后修饰,蛋白质折叠和分子伴侣类的单基因簇较多,有3 105个;信号传导机制类单基因簇最多,达3 330个。以上结果表明单基因簇涉及的COG功能类别较为丰富。
2.2.4 单基因簇的KEGG分析 参考KEGG数据库,对巴哈雀稗单基因簇的代谢途径进行统计分类,结果如图4所示,巴哈雀稗单基因簇可以分为细胞进程、环境信息进程、遗传信息进程、新陈代谢和生物系统共5个主要的代谢途径,包含31类代谢通路;其中,信号转导、翻译、碳水化合物代谢、“折叠、分类和降解”、 代谢概况、“运输和代谢”、氨基酸代谢、能量代谢和脂类物质代谢等通路涉及的单基因簇较多。
結合KEGG pathway数据库,将巴哈雀稗单基因簇定位到具体的183个代谢途径分支上,结果如表3所示,其中甜菜红色素生物合成的基因最少,仅有1个;占总体比例最大(5.54%)的是参与核糖体代谢途径的基因,有1 022个;碳代谢途径的基因占总体的3.84%,有709个;氨基酸生物合成途径的基因占总体的3.14%,有580个。
2.3 SSR预测
通过SSR位点搜索,如表4所示,在巴哈雀稗的99 235个单基因簇中共检测到12 062个SSR位点;其中,三核苷酸重复所占比例最高,为52.85%;六核苷酸重复比例最低,仅占0.59%。在所有检测到的SSR中,出现频率最高的3类基序为:A/T(3 602个)、AT/GA(550个)、AG/CT(487个)。在上述SSR特征分析中,单核苷酸至六核苷酸重复类型均存在,这有助于巴哈雀稗及其雀稗属植物开展遗传图谱构建、基因组差异分析及通用性标记开发等研究,同时也表明了巴哈雀稗的SSR类型较为丰富。
3 讨 论
Illumina高通量测序技术具有成本低、速度快、数据量大、效率高的特点[6],尤其适用于类似巴哈雀稗这样缺乏基因组信息的植物转录组学研究。该研究对高通量测序获得的42 844 132个巴哈雀稗reads序列进一步拼接组装后获得99 235个单基因簇,长度在201~10 795 bp范围内,平均长度为641.96 bp,N50为1 024 bp。 N50是评价组装序列完整性的重要指标,N50越长,代表组装的完整性越好。此次序列组装的数据已满足转录组分析的基本要求,这与贾新平等对海滨雀稗[7]和鸟巢蕨[8]的研究结果类似。
该研究发现巴哈雀稗获得的单基因簇与Nr数据库比对存在53 886个相似序列,同时还获得了45 349个新的单基因簇 (占总体的45.70%),这说明对于缺乏基因组、EST及蛋白序列信息的植物,采用高通量测序技术不失为发现其功能基因的一种有效手段[9]。
功能注释分析发现,基于GO数据库对巴哈雀稗单基因簇相关功能的注释信息尚未完善,还存在部分的单基因簇没有被赋予可能的GO条目,究其原因可能是GO数据库结构设计上存在缺陷,同时还有部分基因的特征尚未被注解,使得这些基因的注释信息并不完整;也可能是这些序列不是注释蛋白质的重要同源物的缘故[10-11]。因此,后期还需要通过其他的方法对单基因簇功能注释进行补充和完善。在KEGG的代谢途径中,部分代谢途径与次生代谢物质的生物合成及信号传导途径有关,譬如β-丙氨酸代谢(ko00410)、苯丙素生物合成(ko00940)、淀粉与蔗糖代谢(ko00500)及丙酮酸代谢(ko00620)等。这些次生代谢产物及信号传导途径均为植物体内重要的抗逆途径,可以缓解或抵御因缺水、水涝等逆境造成的活性氧损伤[12]。从基因组水平上利用COG数据库搜索直系同源体,既可预测未知ORF的生物学功能,又能对巴哈雀稗单基因簇的基因功能进行分类,这能显著提高基因功能注释的准确性。
SSR分子标记具有遗传信息量大、重复性好、多态性丰富、共显性遗传等多个优点[13]。该研究发现的12 062个SSR位点出现频率高且类型丰富,恰好能为下一步巴哈雀稗SSR分子标记开发奠定基础。
该研究采用高通量测序技术对巴哈雀稗幼苗叶片进行了序列组装、功能注释、代谢途径等分析,并获得了其转录组数据,该数据在今后有关雀稗属植物的分子生物学研究中可作为雀稗数植物基因组的参考序列,提供可靠的数据资源。
参考文献:
[1] 侯晓龙,蔡丽平,韩 航,等. 铅胁迫对百喜草叶绿素荧光特性及酶活性的影响[J]. 草业学报,2017,26(3):142-148.
[2] 刘华荣,龙忠富,邓 蓉,等. 百喜草在退耕坡地种植中的水土保持效应及养羊效果[J]. 贵州农业科学,2012,40(7):145-148.
[3] 龙忠富. 百喜草种子生产技术研究[D]. 贵阳:贵州大学,2006.
[4] 黄玉兰,殷奎德,向君亮. 薏苡幼苗叶片转录组分析[J]. 农业生物技术学报,2017,25(3):386-396.
[5] Middleton A M,?beda-Tomás S,Griffiths J,et al. Mathematical modeling elucidates the role of transcriptional feedback in gibberellin signaling[J]. PNSA,2012,109(19):7571-7576.
[6] 牛俊奇,苏建睦,王小敏,等. 基于高通量测序的金钗石斛叶转录组数据分析[J]. 分子植物育种,2018,16(3):747-756.
[7] 贾新平,孙晓波,邓衍明,等. 鸟巢蕨转录组高通量测序及分析[J]. 园艺学报,2014,41(11):2329-2341.
[8] 贾新平,叶晓青,梁丽建,等. 基于高通量测序的海滨雀稗转录组学研究[J]. 草业学报,2014,23(6):242-252.
[9] Yan H,Jia H,Chen X,et al. The cotton WRKY transcription factor GhWRKY17 functions in drought and salt stress in transgenic Nicotiana benthamiana through ABA signaling and the modulation of reactive oxygen species production[J]. Plant Cell Physiol,2014,55(12):2060-2076.
[10] 王伟科,宋吉玲,闫 静,等. 秀珍菇转录组测序和初步分析[J]. 南京农业大学学报,2019,42(2):292-299.
[11] 赵 胡,唐开静,范小莹,等. ‘黑油椿香椿嫩芽高通量转录组测序及萜类代谢物质初步分析[J]. 园艺学报,2017,44(11):2135-2149.
[12] 张少平,邱珊莲,郑云云,等. 紫色黄秋葵转录组功能基因测序及分析[J]. 核农学报,2017,31(4):643-653.
[13] 杨 旭,杨志玲,谭 美,等. 厚朴转录组特征分析及EST-SSR标记的开发[J]. 核农学报,2019,33(7):1318-1329.
(责任编辑:成 平)
