克罗诺杆菌分子分型及毒力机制研究进展
2019-12-03杨捷琳
闫 瑞,钮 冰,杨捷琳*
(1.上海大学生命科学学院,上海 200444;2.上海出入境检验检疫局,上海 200135)
克罗诺杆菌属(Cronobacterspp.),原阪崎肠杆菌(Enterobacter sakazakii),隶属于肠杆菌科,由7 个种和3 个亚种组成[1-3]。属内菌株不同分型种类与临床相关性存在较大差异,其中多数临床分离株为阪崎克罗诺杆菌(C. sakazakii)和丙二酸盐阳性克罗诺杆菌(C. malonaticus)。据报道,多数菌株易感染婴幼儿和老年人等免疫力低下人群,可导致严重疾病(坏死性小肠结肠炎、败血症和脑膜炎),且致死率非常高(40%~80%)[4-6]。随着克罗诺杆菌在不同食品尤其是婴幼儿配方奶粉中被检出,婴幼儿配方奶粉(powdered infant formula,PIF)的污染因与疾病爆发密切相关而受到食品和卫生监管部门的高度重视[7-8]。
至今,分类学依旧是关于克罗诺杆菌研究的主题,因为准确的细菌分类对于可靠的监管控制至关重要,而准确的细菌分类则依赖于有效的分型方法。同时,无论是克罗诺杆菌的流行病学研究还是分子溯源方法的建立都必须基于对目标生物多样性的透彻理解。尽管研究人员已从食品、环境和临床上分离到大量菌株,也对其分类有了较为清晰的认识;但是随着越来越多克罗诺杆菌全基因组测序的完成,研究发现过去基于单个或多个基因的分型方法误将许多无关菌株划分到了一起[9-10]。因此利用全基因组测序数据并采用合适的分型方法来准确地鉴定和快速识别不同来源的克罗诺杆菌显得尤为重要。此外,在克罗诺杆菌流行病学与毒力机制研究方面发现,菌株间致病性存在较大差异,毒力具有多样性,致病机制复杂多变[11-14]。进一步深入研究克罗诺杆菌属内各菌株的致病机制,从分子角度阐明相关致病机制和毒力特征,并选用合适的动物模型对该属菌株进行毒力验证性实验是当前克罗诺杆菌研究的主要方向之一。
1 克罗诺杆菌分型方法
克罗诺杆菌的分型方法对于克罗诺杆菌的分类学研究起到至关重要的作用。从最初的Farmer[15]、Iversen[16]等采用表型分型方法建议将黄色阴沟肠杆菌改名为阪崎肠杆菌,并依据生化谱将实验菌株划分为15 个生物群,后扩展至16 个生物群;再到2007年—2008年Iversen等[1-2]采用多种新的分型方法(DNA-DNA杂交、核糖体分型和全长16S rRNA基因序列分析等)建议建立一个新属即克罗诺杆菌属代替先前的单一物种阪崎肠杆菌。这一新属当时由5 个新种、1 个基因种(Cronobacter genomospecies1)组成。直到Joseph等[3]进一步结合基于7 个管家基因的分型方法建议将C. genomospecies1命名为C. universalissp. nov.(尤尼沃斯克罗诺杆菌新种),其同时发现了一个新的种C. condimentisp. nov.(康迪蒙提克罗诺杆菌新种),最终形成了包含7 个种的克罗诺杆菌属。克罗诺杆菌分类地位变化并不仅仅依赖于1 种新的分型方法,而是多种传统分型方法与新分型方法的结合应用(表1)。这些分型方法不仅为克罗诺杆菌的准确分类提供了依据,同时也为克罗诺杆菌的分子溯源及毒力机制研究奠定了基础。
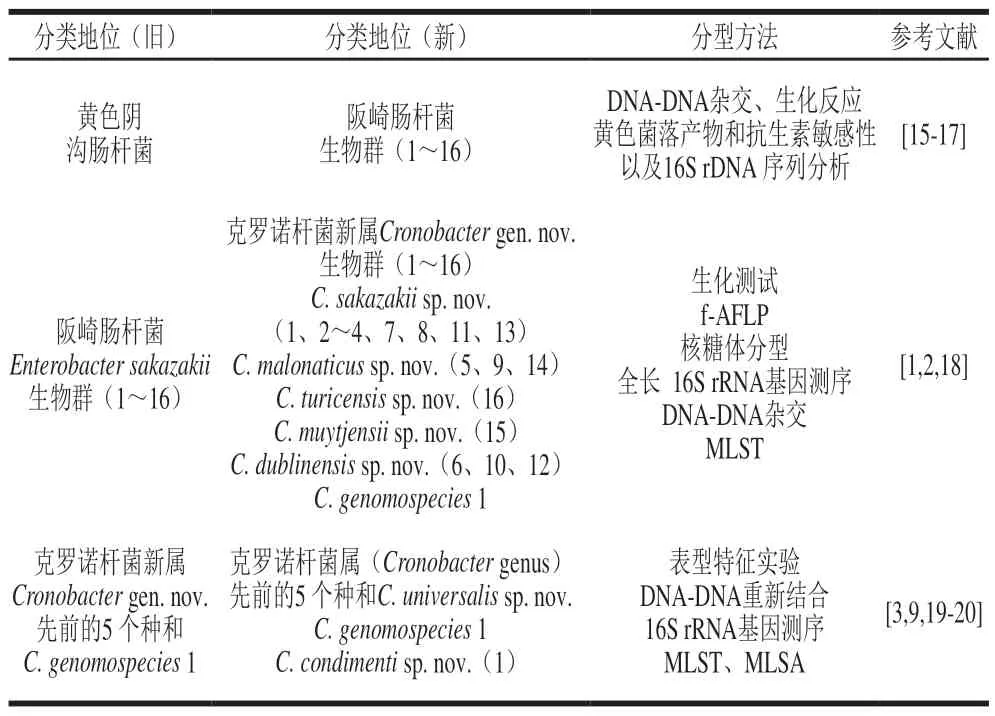
表 1 克罗诺杆菌分类地位变化及分型方法Table 1 Changes in taxonomic status of Cronobacter and typing methods
1.1 生物分型
最初,Farmer等[15]采用DNA杂交、生化测试、黄色菌落产物以及抗生素敏感性等系列方法对57 株阪崎肠杆菌进行分类实验,建议将“黄色阴沟肠杆菌”更名为阪崎肠杆菌,并建议将其另立为肠杆菌属下的一个新种。同时基于10 个生化测试定义了15 个克罗诺杆菌生物群,并用于菌株的形态鉴定和区分。然而,基于DNA序列的方法所描述的菌株形态表明,这种生物分型方法存在严重缺陷,仅有不超过50%的菌株被正确分配到克罗诺杆菌[16]。因此,生物分型方法对于菌株的形态鉴定及准确定义被认为是不可靠的。此外,生物群与单独的克罗诺杆菌种之间也没有准确的相关性。尽管生理生化分型方法存在较多弊端,但因其普遍的适用性和基础的鉴定能力,可作为经典的分析方法。目前研究人员主要利用API 20E生化鉴定系统来进行生化分型,该方法基于多种不同的生理生化指标能够将克罗诺杆菌鉴定至属的水平,若要鉴定至种的水平则需要进一步增加生理生化实验。
1.2 脉冲场凝胶电泳分型
早期在对克罗诺杆菌感染的流行病学分析中,使用两种限制性酶(XbaI和SpeI)的脉冲场凝胶电泳(pulsed-field gel electrophoresis,PFGE)是最常用的溯源分型方法[6,17,21]。常用于奶粉和婴幼儿配方奶粉中微生物污染源的追踪[22-25],该方法稳定、可重复性强,应用基础广泛扎实,但该方法仍存在不能鉴定菌株和无法确定菌株的相关性;另外,由于固有DNA酶的活性使得某些菌株不能生成具有明显特征的PFGE图谱,故不能达到准确区分菌株的目的。因此,PFGE方法正在被逐步取代,全世界的疾病预防控制中心都正在转向使用全基因组测序方法作为国际食源性疾病监测分子分型网络(https://www.cdc.gov/pulsenet/index.html)监测的基础,同时推广基于全基因组序列的分型方法[26-27]。
1.3 单基因分型
基于序列的细菌鉴定方法是从单基因座测序开始的,通常能够鉴定至种水平。如克诺罗杆菌的16S rRNA、ompA、dnaG、rpsU、cgcA、fliC、rpoB和fusA基因分型[19,28-30]。其中16S rRNA基因因其种间的高度相似性(97.8%~99.7%)而存在一定局限性[20]。2008年,Iversen等[2]发现C. malonaticussp. nov.并不是C. sakazakii的亚种,主要是因为16S rRNA序列分析不能将这两个种区分开,但DNA-DNA杂交实验显示这两个种的DNA相关性小于70%,所以把生物群1~4、7、8、11和13归为阪崎克罗诺杆菌,生物群5、9和14划分到丙二酸盐阳性克罗诺杆菌。此外,后续针对克罗诺杆菌中的ompA、dnaG和rpoB等基因设计的分型方法均存在类似的局限性,一部分费时耗力,一部分则未经过涵盖克罗诺杆菌7 个种大量菌株的分型验证[31]。其中,fusA等位基因与克罗诺杆菌的系统发育一致,可用于克罗诺杆菌属菌种的鉴定。该方法能够成功地将克罗诺杆菌的7 个种区分开,但对于克罗诺杆菌属同种菌株间的研究则存在较大限制。
1.4 O-血清分型:gnd和galF基因分析
早期的血清分型方法是使用在动物体内产生的抗体进行血清分型,随着聚合酶链式反应(polymerase chain reaction,PCR)引物可以被设计用于O-血清型(rfb基因座)的PCR扩增,然后对PCR扩增产物进行限制酶消化以产生可识别的条带图案,血清分型便不再需要使用动物。这种方法被许多研究人员用于Cronobacterspp.研究[32-34]。但Sun Yamin等[35]因使用该方法将一些C. malonaticus菌株误认为C. sakazakii,并且错误地确定了C. sakazakii的血清型。其次,可能由于O-血清型中的变体区域发生在PCR引物的目标区域之外,故PCR检测分型并不能覆盖所有C. sakazakii所产生的O-血清型的扩增产物[32-33]。因此O-血清型分型的PCR-引物方法逐渐被位于rfb区域侧面的等位基因gnd和galF的分析方法所取代,这种基于DNA序列的方法用于确定O-血清型则更可靠,操作也更简便,同时增加了克罗诺杆菌中可定义血清型的数量范围(24~34 种)[36]。
1.5 多位点序列分型
MLST主要是将能够反映全基因组系统发育关系的多个看家基因的测序数据与数据库中已登记数据进行比对分析从而获得相应序列型。该方法能够将高度相似的菌株进行有效区分。克罗诺杆菌MLST通常需要7 个管家基因:atpD、fusA、glnS、gltB、gyrB、infB和ppsA。这些基因座最初是用特别设计的引物单独测序的,所需成本非常高。随着下一代测序技术(next generation sequencing,NGS)的发展,使得全基因组测序成本的大大降低,同时促进了克罗诺杆菌基因序列数据库(Pub-MLST)的建立和发展[9,31,37]。Forsythe[9]和Ogrodzki等[10]详细综述了1 600多株克罗诺杆菌的MLST结果。截止2018年11月5日,在数据库中分离菌株已达2 600多株,这些克罗诺杆菌属内菌株的详细测序数据(来源、分布、菌种构成、序列类型和克隆复合物等)和研究进展均可在线获得(http://pubmlst.org/cronobacter/)。
对于克罗诺杆菌属各菌株之间的研究,MLSA已被证实是一个非常有用工具。2009年Baldwin等[18]利用MLSA成功将C. sakazakii和C. malonaticus两个种区分开。此外,MLST显示C. sakazakiiST4菌株为优势菌株(22/60)。2012年Joseph等[19]采用MLST方法对325 株克罗诺杆菌的系统发育关系进一步证实了7 个种的存在,同时发现种内变异从C. sakazakii的低多样性到一些种的广泛多样性,从而推测物种之间可能存在基因转换。此外,还发现C. sakazakii是临床来源的优势菌种,C. sakazakiiST4是脑膜炎病例脑脊液分离株的主要序列类型。2014年Forsythe等[9]通过采用更具辨别力的编码核糖体蛋白基因的多位点序列分析(ribosomal-MLST,rMLST)和基于直系同源基因簇核心基因的多位点序列(clusters of orthologous genes-core gene MLST,COG-cgMLST)分析方法对107 个克罗诺杆菌全基因组数据进行分析,进一步证实C. sakazakiiCC4克隆谱系非常稳健。尽管MLST克隆识别对于克罗诺杆菌病原体的鉴定是有效的,但它对于微生物溯源可能会因相同序列类型(senquence type,ST)当中包含大量不相关菌株(地理来源、分离时间、宿主等)而适得其反[31]。这一点在COG-cgMLST分析结果的ST4聚类中也能够得到印证,其中ST4包含了大量不相关菌株,这或许是该聚类存在大量分枝的原因。另外,这或许也可以解释不相关的临床C. sakazakii菌株却具有相同的PFGE脉冲型这一现象。为了解决这一问题,单核苷酸多态性(single nucleotide polymorphism,SNP)分析和规律成簇的间隔短回文重复序列(clustered regularly interspaced short palindromic repeats,CRISPR)分型在近几年已被应用于相同ST的Cronobacter.spp.全基因组研究中。
1.6 SNP分型
单核苷酸多态性能够反映分离株之间相当大的系统发育距离,Masood等[38]采用基于全基因组序列的SNP分型技术对1994年法国新生儿重症监护中心的C. sakazakii相同ST菌株进行了溯源分析,其结果显示SNP分型方法可以对相同序列型菌株进一步分型,来自不同基因型簇的阪崎肠杆菌分离株具有共感染新生儿的能力,然而爆发来源尚未确定。此外,Guo Qingyan等[39]也通过类似方法对源自乳品的32 株C. sakazakiiST13菌株进行溯源分析,发现虽然部分C. sakazakiiST13菌株表现出地理位置的聚簇性,但整体来看SNP聚簇与原产地及乳制品产品类型之间没有明显的相关性。虽然SNP分型方法具有高度分辨性,但对参考菌株的选取较为敏感,即选取不同参考菌株SNP分型结果存在差异。SNP分析为计算密集型,因此可能分析时间较长。对于分析大型序列集,必须访问高性能计算。此外,应用该方法进行分析需要大量的生物信息学知识[27]。
1.7 CRISPR-cas分型
CRISPR-cas阵列反映了菌株对噬菌体和质粒的暴露,通常,CRISPR-cas系统可具有多达3 个区段:cas基因、阵列上游的富含AT的前导序列、由两个重复序列(24~48 个核苷酸)组成且中间插入长度相似的间隔区的短CRISPR阵列,这些间隔区通常来自移动遗传区,如噬菌体和质粒等。间隔物在引导端依次添加,并且给定的间隔物具有高度特异性。由于对噬菌体和质粒的不同暴露,CRISPR-cas阵列在密切相关的菌株之间可能不同,导致间隔序列的变化。这些基因座可用作分子亚型的替代靶标,并且可提供比MLST(7 个位点)和PFGE更高的菌株分辨率。因此,该方案可能是对于高克隆生物如克罗诺杆菌有用的分型工具[10,31]。Ogrodzki等[40]曾将CRISPR-cas阵列应用于分析C. sakazakii4 个致病型:CC1、CC4、ST8和ST12。结果显示该方法能够根据菌株的间隔基因阵列将同一ST中的菌株进行区分。此外,他们进一步将该方法应用于克罗诺杆菌其余6 个种相同序列型菌株的研究,并证明了CRISPR-cas分析用于流行病学目的有用性。
随着克罗诺杆菌全基因组测序数据的增加,一方面使得Pub-MLST数据库中菌株ST、CC数据大规模增加;另一方面也使得对大量相同序列型菌株进行更全面的全基因组比较分析成为可能。这不仅有助于克罗诺杆菌属内相同ST菌株的进一步细分,而且有利于大量的毒力因子的揭示,从而为该菌的毒力机制研究奠定基础。
2 克罗诺杆菌毒力机制
最初克罗诺杆菌因与新生儿感染相关而被人们所关注,现已被公认为主要引起成人感染。克罗诺杆菌属的7 个种可根据其临床相关性进行分组:第1组包括C. sakazakii和C. malonaticus,构成了所有年龄组的大多数临床分离株;第2组包括C. turicensis和C. universalis,很少被报道;其他3 种C. dublinensis、C. muytjensii和C. condimenti主要是环境共生物,可能很少或没有临床意义。因此,克罗诺杆菌毒力机制的研究主要集中于C. sakazakii和C. malonaticus[9,12,36,41-45]。其中,与特定新生儿和成人感染相关的致病型分别是:C. sakazakiiCC4、ST12与新生儿感染有关,C. malonaticusCC7与成人感染相关。其他的潜在致病性研究,如分离自婴幼儿配方奶粉的C. sakazakiiST83菌株通过感染斑马鱼的实验结果显示其致死率非常高(90%~100%)[14]。过去,开展动物实验通常选用的菌株为C. muytjensiiATCC 51329T,这是前阪崎肠杆菌物种的PreceptrolTM菌株[46]。然而,鲜有报道关于该物种的临床病例,因此研究的相关性尚不确定。近期也有研究人员通过采用全基因组测序的方法针对代表性菌株如C. sakazakiiBAA-894进行致病性和毒力特征的深入研究。
2.1 流行病学
由于发达国家和一些发展中国家的报告标准不同,甚至会出现漏报的情况,造成感染病例较少、病例报告不足的现状,以至于目前克罗诺杆菌的流行病学仍是不完整的、模糊不清的。现已知患病新生儿的主要症状有坏死性小肠结肠炎、败血症和脑膜炎。前者是非侵入性的,而在败血症和脑膜炎中,病原体可能通过肠上皮层细胞附着并侵入宿主。同时,关于C. sakazakiiCC4在新生儿脑膜炎病例中占主导地位的原因尚不清楚,这可能与该克隆谱系菌株的环境适应能力较强及潜在的毒力因子有关。C. sakazakiiCC4菌株已从婴儿配方奶粉[22,24,47-50]和多数国家的奶粉生产厂中分离出来,因此可能代表一种稳定持久的变异菌株,导致了新生儿感染的增加。C. sakazakiiST12与坏死性小肠结肠炎病例有关而非新生儿脑膜炎或败血症[38]。需要注意的是,NEC是新生儿常见的多因素胃肠道疾病,它不单与Cronobacterspp.有关,可由多种细菌病原体引起。关于成人的克诺罗杆菌感染报道指出[9,44],C. malonaticusCC7似乎与成人病例相关。成人的克罗诺杆菌感染表现出多种症状:结膜炎、胆汁性败血症、尿脓毒症、阑尾炎、伤口感染和肺炎。可能由于血脑屏障的成熟,成人病例较少发生败血症和脑膜炎。另外,成人感染的来源尚不明确,发病率增加的原因也尚不清楚。
2.2 致病性和毒力因子
目前关于克罗诺杆菌感染所引发的高死亡率的详细机制仍然知之甚少,其中阪崎克罗诺杆菌主要毒力因子和感染致病机制模型可参见图1[12]。
如图1所示,该致病菌能够编码多个与致病过程密切相关的毒力因子,这些毒力因子在包括黏附于宿主表面、穿过、侵入和破坏肠上皮细胞内的肠屏障等致病过程中起到了重要的作用。据报道,阪崎克罗诺杆菌不仅能够从宿主细胞顶端侵入,而且也能够从基底外侧侵入,从而转移到大鼠更深的器官(脾脏和肝脏)[51-52]。由于克罗诺杆菌的致病研究还比较分散,没有形成系统性。因此,克罗诺杆菌毒力因子列表仅列举了部分与发病机制相关的主要毒力因子(表2)。

图 1 假设的阪崎克罗诺杆菌感染和发病机制模型[12]Fig. 1 Proposed model for Cronobacter sakazakii infection and pathogenesis[12]
2.3 外膜蛋白
克罗诺杆菌可以侵入人体肠道细胞,在巨噬细胞中复制,并侵入血脑屏障[51-52]。体外研究表明,克罗诺杆菌在对哺乳动物肠细胞的附着和侵袭,巨噬细胞中的存活率及对血清的抗性方面与E. cloacae(阴沟肠杆菌)和Citrobacter freundii(弗氏柠檬酸杆菌)相当,但是低于Salmonella typhimurium(鼠伤寒沙门氏菌)。外膜蛋白A(OmpA)和外膜蛋白X(OmpX)可能在克罗诺杆菌穿透血脑屏障中起作用,尽管导致脑细胞破坏的机制尚不清楚,并且部分可能是宿主反应[53-54]。C. sakazakii产生的外膜囊泡,可能会导致宿主细胞产生细胞病变作用[55]。近期,也有研究人员通过基因芯片技术来研究与外膜蛋白致病密切相关的外膜囊泡。结果显示,外膜囊泡可能参与外膜蛋白的包装和转运过程[56]。
2.4 唾液酸的利用
C. sakazakii和一些C. turicensis菌株可以利用外源唾液酸作为生长的碳源,这或许具有临床意义。这可能是一个主要的进化宿主适应机制,因为唾液酸存在于母乳、黏蛋白和神经节苷脂中[57]。由于唾液酸与大脑发育相关,因此其也是婴幼儿配方奶粉的一种成分。C. sakazakii也能够在神经节苷脂GM1上生长,其中唾液酸是唯一的碳源[58]。
2.5 质粒携带的毒力因子
质粒携带的毒力因子是一种重要的潜在毒力因子,如克罗诺杆菌纤溶酶原激活物(Cronobacterplasminogen activator,CPA),仅在C. sakazakii和C. universalis中编码一种外膜蛋白酶[59]。另外,Eshwar等[13]进行了以斑马鱼为动物模型的克罗诺杆菌感染实验,他们的实验不仅证实了RepF1B样质粒在克罗诺杆菌中作为“毒力质粒”的作用,同时巩固了两种推定的毒力因子cpa和zpx在体内发病机制中的重要性。此外,Chase等[14]通过采用全基因组测序和系统发育分析的方法发现C. sakazakiiH322和其它ST83菌株都存在pESA3-样毒力质粒,其中一株存在pESA2-样毒力质粒,同时还发现了pCS1-样毒力质粒。这些结果表明ST83菌株可能具有高的致病性。

表 2 克罗诺杆菌主要的已知毒力因子特征Table 2 Characteristics of major known virulence factors of Cronobacter
2.6 其他潜在毒力因子
克罗诺杆菌的测序基因组揭示了一系列的黏附素,如外膜蛋白、外排系统、铁吸收机制、溶血素和VI型分泌系统等[60-63],这可能有助于进一步全面了解该菌的毒力机制。其他候选毒力决定因素包括用于巨噬细胞存活的超氧化物歧化酶、鞭毛、金属蛋白酶、肠毒素[52,64-67]。如何选取合适的新生动物模型对上述潜在毒力因子进行有效验证是当前克罗诺杆菌毒力机制研究的主要方向之一。
3 结 语
从克罗诺杆菌的研究历程来看,克罗诺杆菌的分类地位随着分型方法的改进在不断的发生着变化,直到2012年克罗诺杆菌才形成较为明确的分类。近几年的研究则主要基于这一分类基础,进一步对不同种菌株进行MLST研究,然而通过全基因组测序和比较基因组分析发现Pub-MLST数据库已登记的MLST结果中因包含了大量不相关菌株而很难将这些分型结果应用于克罗诺杆菌的溯源分析[9-10,30]。同时,克罗诺杆菌分类形成之前的一些关于阪崎肠杆菌的毒力机制研究则需进一步的验证。本文着重介绍的分型方法,也是当前全基因组分型中较为常用的分析策略。其中API 20E可作为克罗诺杆菌属的初步鉴定,而PFGE正在被逐步取代转向基于全基因组序列的分型方法。基于单基因如fusA的分型可用于种水平的鉴定,其他单基因分型则有助于对克罗诺杆菌功能及毒力基因的研究。进一步采用基于gnd和galF基因及MLST分型则可获得更多关于菌株血清型,ST和CC型信息,从而能够进行系统发育关系、菌株致病相关性、流行病学等研究。在此基础之上从全基因组层面对相同ST克罗诺杆菌进行深度解析如COG-cgMLST、SNP和CRISPR-cas分析,则可为确定菌株来源、获取更多与菌株流行病学特性及毒力因子相关信息奠定基础。克罗诺杆菌的毒力机制研究尚缺乏统一结论,研究较为分散。一方面需要我们不断完善其流行病学信息,从而获得更多确切的致病信息;另一方面随着基因分型的发展,菌株分类更加清晰,先前与毒力机制相关的一些结果可能存在缺陷需要再次验证。综上所述,结合克罗诺杆菌Pub-MLST数据库从全基因组层面对属内不同来源菌株进行剖析,选取合适的新生动物模型对基因测序揭示的毒力因子进行验证,不但可以促进该菌的溯源研究,而且对于详细阐明其毒力机制有着深远影响。
