保留非全长读段的ISO-seq数据转录组表达分析
2019-09-06刘学军瞿锡垚
刘学军 瞿锡垚 张 礼
(1.南京航空航天大学计算机科学与技术学院,南京,211106;2.南京林业大学信息科学技术学院,南京,210037)
引 言
由于选择性剪切(Alternative splicing,AS)的存在,一条前体mRNA可以剪切出多条mRNA并指导蛋白质的生成,这种转录组一对多的剪切方式是造成生物多样性的最重要原因之一。而各项研究都表明选择性剪切现象普遍存在于高等真核生物中[1],同时一些特异剪切方式产生的新型异构体也是导致基因疾病的重要原因之一,因此研究选择性剪切对于揭示人类疾病机制具有重要的意义。
基因及异构体表达水平计算是研究选择性剪切的一种重要途径,具有高通量特性的第二代测序技术RNA-seq拥有量化转录片段的突出优势,在这一领域具有较多有效的应用[2-3],很多方法采用RNA-seq数据计算基因以及异构体的表达水平。例如,基于泊松分布的PGseq[4]和NURD(Non-uniform read distribution)[5],以及基于读段产生式的Cufflinks[6]和RESM[7-8]等。但是由于读段长度短、GC误差等缺点的存在使得RNA-seq技术在识别全长异构体方面显得十分乏力;另外,在表达水平计算方面,RNA-seq技术也面临着读段多源映射的问题(参考序列中大量重复和同源序列的存在,导致一个读段映射至多个位置)[9],在已有的大多数方法中,都存在严重依赖注释信息的情况,而注释信息不完善降低了表达水平计算的准确度。由于统计每个外显子上读段数量是基于RNA-seq技术计算基因及异构体表达水平的基础[10],因此即使采用相同注释信息的模型,不同方法在估计表达值时也存在较大差异[11]。
近几年诞生的第三代测序技术以其读段长度长、无聚合酶链式反应(Polymerase chain reaction,PCR)过程引入的误差等特点,迅速得到研究者的关注,并应用于RNA-seq技术不适用的场景。ISO-seq技术是PacBio公司开发的用于转录组研究的第三代测序技术,该技术从细胞中分离出mRNA,在size-selection(长度选择)之后制备成ISO-seq文库,用于测序仪测序。整个测序过程没有对测序片段作任何打断处理,这样的测序结果可认为是测序片段的完整读段,因此ISO-seq技术在诞生之后,被大多数研究者应用于转录组重构和基因组组装等领域[12-13]。但该技术的测序结果存在较高的错误率,为了解决这一问题,大多数方法同时使用RNA-seq和ISO-seq数据。一方面利用RNA-seq数据的准确性进行ISO-seq数据纠错,另一方面利用RNA-seq数据的高通量辅助预测异构体并计算异构体达水平。例如,IDP[14]方法是ISO-seq数据处理的代表方法,它使用混合策略,以聚类方式从RNA-seq数据和注释库中找出junction(外显子剪切点),去除没有junction支持的ISO-seq长读段,将得到的非冗余多外显子长读段作为预测异构体,再将RNA-seq数据比对至预测异构体并计算各个预测异构体的表达水平。
IDP在除去冗余全长读段时,不仅去掉了信息一致的全长读段,还删除了全长读段之间的包含情况,即当一条较短的全长读段包含于一条较长全长读段时,只保留较长的读段。但从ISO-seq测序技术原理来看,在制备cDNA文库时并没有进行类似RNA-seq测序技术的随机打断,因此本文认为一条全长读段等价一个异构体,去掉包含关系的读段会遗漏异构体,最终影响到异构体的预测结果。另外,大多数研究工作认为ISO-seq数据的通量低,不适合计算表达水平,其主要原因在于这些研究工作大都丢弃了占到所有数据50%~60%的非全长读段[15]。而非全长读段的产生是由于测序时酶失活,导致测序过程无法继续进行,但非全长读段仍然具有ISO-seq数据超长读长的特性,也包含外显子信息,能够反映样本中转录本的浓度,丢弃非全长读段将会直接影响基因和异构体表达水平的计算,故目前的方法大多采用ISO-seq数据和RNA-seq数据相结合的方式进行异构体表达水平的计算。
本文首次提出仅利用ISO-seq数据,且保留非全长读段进行基于狄利克雷采样的探测与预测(Dirichlet sampling for isoform detection and prediction,DSIDP)方法。同时,第三代测序技术虽然拥有超长读长测序,但也无法保证全长读段数据涵盖所有表达异构体,针对一些没有全长读段数据的异构体预测问题,本文在沿用DSIDP预测异构体思想的基础之上,还提出了一种基于马尔科夫链的异构体探测与预测(Markov chain for isoform detection and prediction,MCIDP)方法。两种模型均在模拟数据集和真实数据上得到了有效验证。
1 实验方法
1.1 数据特性
图1显示了ISO-seq数据中全长读段和非全长读段长度分布直方图,图中数据来自PacBio公司公开数据集 MCF-7(http://www.pacb.com/blog/data-release-human-mcf-7-transcriptome/)。本文统计了 6个cell的原始数据(如表1所示),其中按照ISO-seq技术的size-selection原则,对样本长度1~2 Kb,2~3 Kb和>3 Kb三个范围的Cell各选取两个。从统计结果可以看出全长读段和非全长读段的长度分布具有相似的模态,数据多集中在长度为1~3 Kb的区间内,这说明非全长读段数据也具有远超过RNA-seq数据的长度,从第三代测序数据超长读长这一本质特征来说,非全长读段与全长读段一样,也包含关于异构体的有效信息。并且随着样本序列长度的增加,非全长读段也随之增加,并达到接近60%。如果在异构体的构建中不考虑这部分数据,相当于丢弃了大部分实验数据。因此将ISO-seq数据应用于转录组学研究领域时,保留非全长读段具有重要意义。

图1 ISO-seq数据中读段长度分布直方图Fig.1 Histograms of ISO-seq reads

表1 MCF-7数据集读段统计Tab.1 Read statistics of MCF-7 data set
为了进一步说明ISO-seq数据用于计算表达水平的可行性,随机选择了4个基因并统计它们在ISO-seq数据和RNA-seq数据中外显子上读段数分布情况,结果如图2所示,第一行显示了RNA-seq数据结果,第二行显示了ISO-seq数据结果。从图2中可以看出ISO-seq数据与RNA-seq数据具有极为相似的分布模态,这说明ISO-seq数据与RNA-seq数据一样,能通过读段计数反映出样本中相应转录本的浓度,进而可以用来计算转录组的表达水平。
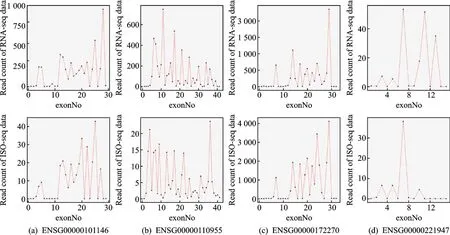
图2 4个基因在ISO-seq数据和RNA-seq数据中外显子上的读段分布Fig.2 Distribution of reads on exons of four genes in ISO-seq data and RNA-seq data
1.2 原始数据处理
ISO-seq的下机数据中全长和非全长读段是混合在一起的,需要按照一定的准则将其区分开。PacBio公司提供的SMRT Analysis软件根据读段数据两端是否均存在接头序列,将其分为全长读段和非全长读段,但这样的方式需要原始读段数据之间的比对,对计算效率影响较大。图3展示了PacBio测序原理,ISO-seq数据的cDNA文库是两端接上接头的哑铃状结构,测序时会在整个结构上循环进行[16]。根据这样的测序原理,本文使用一种简单高效的方法区分全长和非全长读段。当一个零膜导波管(Zero mode waveguide,ZMW)中出现多条Subread时,从中选择最长的读段划分至全长读段集合,否则,将唯一的一条Subread划分至非全长读段集合。整个过程不仅区分出了全长和非全长读段,还去掉了多条Subread中的冗余读段。
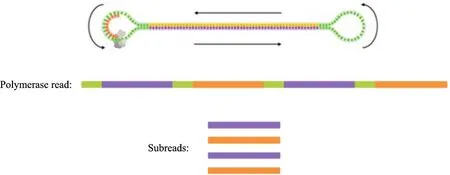
图3 PacBio测序原理Fig.3 PacBio sequencing principle
ISO-seq数据的另一特征是较高的测序错误率,目前的大多数研究工作均使用RNA-seq数据对其进行纠正,本文使用LoRDEC[17]对非冗余读段(包括全长读段和非全长读段)进行纠错处理,参数值k设置为21,s设置为3。纠错后的数据使用BWA-MEM[18]比对至参考基因组,借助基因注释库信息从比对结果中获取到读段中的外显子序列。对于没有RNA-seq数据的情况,同样可以采用仅使用ISO-seq数据进行自纠错的方法,例如Chen等[19]使用最长的读段作为种子来收集其他所有读段,构建高度准确的读段数据。
1.3 真实数据有效性验证
MCF-7数据集中共有119个Cell,且测序时间并不都是一致,因此本文在建模之前验证了数据的有效性。选取6个Cell的真实数据,分为两组,每组均包含Size-selection的3个长度范围,即Cell 1,Cell 3,Cell 5为一组记为Group 1,Cell 2,Cell 4,Cell 6为另一组记为Group 2。对两组数据中的5 665个公共基因通过计算RPKM[20]值得到表达水平,在对数刻度上验证两组数据获得的基因表达水平的吻合性。结果如图4所示,相关系数为0.900 6。可以看出这两组重复实验在基因表达值上具有很高的一致性,表明多次测量得到的读段具有较好的可重复性,因此数据集的有效性得以验证。

图4 MCF-7数据集重复实验结果对比Fig.4 Comparison of repeated experiment results of MCF-7 data set
1.4 DSIDP模型
PacBio测序技术从细胞中提取到mRNA后没有进行分子随机打断,因此本文认为经过对下机原始数据的处理后,得到的所有非冗余全长读段数据即为细胞中表达的异构体集合,并将这个集合作为模型的异构体预测结果。
借鉴RNA-seq数据计算表达水平时统计外显子上读段数量的方式,本文将所有ISO-seq读段数据映射至异构体预测集,并统计各预测异构体上读段数量,在总读段数上做归一化得到其表达水平。映射过程中,ISO-seq数据也将面临相同基因下多个异构体之间的多源映射问题。MCF-7数据集的6个Cell中,35%的读段存在多源映射的情况,这远低于二代数据70%读段的多源映射情况[9]。这里的ISO-seq数据多源映射是指一条非全长读段映射至多条预测异构体,如何分配这样的非全长读段是计算异构体表达水平中要解决的核心问题。为了解决这个问题,本文提出了DSIDP模型。DSIDP是一个基于Dirichlet分布,对该问题进行建模求解的算法,使用随机采样方法将发生多源映射的非全长读段映射到概率最大的异构体,通过这样的方式利用非全长读段进行异构体表达比例的计算。具体算法过程如下:
算法1DSIDP
输入:全长读段数据XFL,非全长读段数据XnFL以及异构体预测集合T,和均代表一条读段数据,Ti代表一个异构体,|T|=k;
输出:预测异构体的表达水平向量E,每一维代表相应预测异构体的表达水平。
(1)将读段数据矩阵XFL和XnFL映射至异构体预测集合矩阵T,统计每个异构体上唯一映射读段数量,得到一个k维向量,并对每一维在所有维度上做归一化记为τ。
(2)将发生多源映射的读段数据合并记为Xm,则每一个均对应一个tj(tj⊆T)和一个τj(τj⊆τ),τj是归一化的结果,Isoform~Dirichlet(τj)。
(3)从Isoform~Dirichlet(τj)中采样得到变量isoform,其中各维度上的概率值表示属于对应异构体的可能性,选择概率最大的异构体,在其读段计数上加一。
(4)遍历完所有Xmj之后得到新的异构体读段计数向量,归一化处理结果记为表达水平E。
1.5 MCIDP模型
在RNA-seq数据的处理中,LeGault等[21]使用概率连接图(Probabilistic splice graphs,PSGs)方法对异构体结构进行预测。在固定基因结构的情况下,量化基因的选择性剪接事件,从测序数据中找到异构体的junction,通过junction之间的跳转做出异构体结构的预测。本文将这样的思想运用到第3代测序数据中,提出了MCIDP模型。由于ISO-seq数据长读段的特点,在junction跨越很长的区域时,也能有读段的支持,因此这样找到的junction较之LeGault等使用RNA-seq数据要更为精确和全面。
MCIDP使用马尔科夫链对异构体junction之间的跳转进行建模。一个基本的马尔科夫链包含3元素:状态节点(V)、初始状态概率向量(π)、状态转移概率矩阵(A),因此,模型可以表示为G=(V,A,π)。其中状态节点由基因结构决定,将基因外显子由编号从小到大进行排列,并在该排列的两端加上起始点V0=0和终止点VM=M(M=|V|-1),即为状态节点集合;Aij表示状态节点i转移至状态节点j的概率值,且有Aij∈[0,1],∀i,;πi表示由状态节点i作为路径起始点的概率值,且有从模型建立的整个过程可以看出,MCIDP方法只需要知道基因外显子组成,不依赖注释库中异构体的注释信息。图5为MCIDP建模示意图。
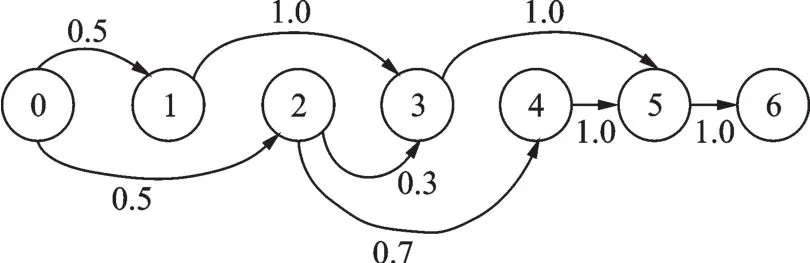
图5 MCIDP建模示意图Fig.5 Modeling diagram of MCIDP
模型的一条路径σt即代表一个可能存在的异构体,例如图5中的路径V0V1V3V5V6。在任意一条没有全长读段的路径上,如果存在其他的读段能够拼接出该路径,那么该路径也能够被模型模拟并预测。表示路径t中的第k个状态节点,根据之前的设定,=V0,=VM,其中e=|σ|,表示路径所包含的状态节点数,下标指示路径节点,上标指代路径。路径中转移概率的累积乘积Expr(σt)可表示为

使用极大似然估计出马尔科夫链模型的参数π和A,令Nij表示状态节点i与状态节点j之间的junction总个数,则对于Aij和πi的极大似然估计有

MCIDP沿用了DSIDP从全长读段中建立异构体预测集的思想,将所有非冗余全长读段数据作为模型预测异构体的初始集合。由于构造的图模型中有些路径的junction结构较为相似,可以进行合并计算,所以需对所有其他可能存在的异构体根据定义的距离公式,将其路径概率累加到结构最近的预测异构体中。这里距离公式的定义同时考虑到了两个junction之间局部跨区域的差异和所有junction之间累积起来的全局差异,具体描述如下:
令St表示一个异构体的外显子序列,基因的第i个外显子包含于其中,则=1,否则=0,|St|=M。对于两个异构体外显子序列St1,St2,若<i<M,则认为节点i处存在这两个异构体的相似junction,且为开始位置,若=,i<j<M,则认为节点j处为该相似junction的结束位置。由此,两个异构体中相似junction的初步距离定义为J(St1,St2),可表示为

式中λJ为度量因子,作为指数距离公式中的底数。考虑到差异区域长度对距离的影响,令li表示基因第i个外显子的长度,L(St)表示异构体St的长度,则两个junction的差异区域长度对距离的影响可以定义为Iij(St1,St2),可表示为

式中λI可视为惩罚因子,所以两个异构体中相似junction的最终距离定义为Dij(St1,St2),可表示为

则两个可能存在的异构体的距离D(St1,St2)可表示为
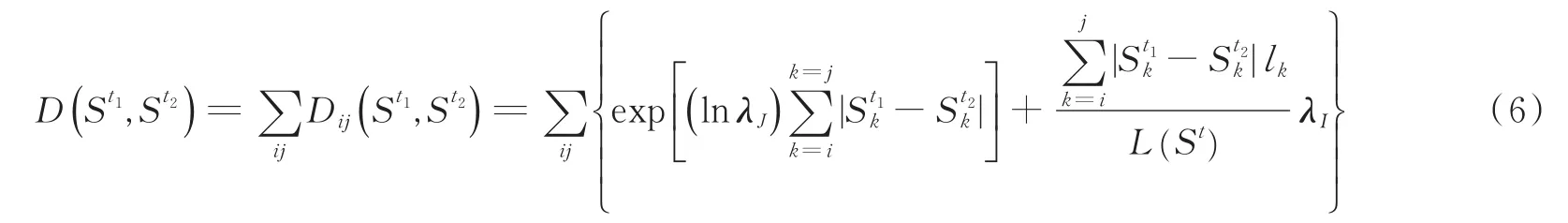
对于超出距离阈值的可能存在的异构体,对其作Kmeans聚类处理,距离公式使用式(6),并将聚类中心作为新的预测异构体添加至异构体预测集合,该预测异构体的表达比率等价于以其为聚类中心的所有可能存在的异构体路径概率之和。最终,模型输出异构体预测集合及集合元素各自的概率值,该概率值即为该基因每个可能存在的异构体的表达比例。
2 实验结果与分析
2.1 实验数据
本文使用了一个模拟数据集和一个真实数据集来验证两个模型的有效性。模拟数据集中,假设了一个拥有10个外显子和4个异构体的基因,并设置异构体的表达比例分别为t1=0.3,t2=0.3,t3=0.2和t4=0.2,如图6所示。按照设定的比例,采样生成了100条全长读段数据,根据后续实验的需求从中随机选取n条全长读段,作随机打断处理,生成非全长读段。真实数据集来自PacBio公开数据MCF-7,本文选取了其中6个Cell的数据,各Cell数据读段的统计情况见表1。
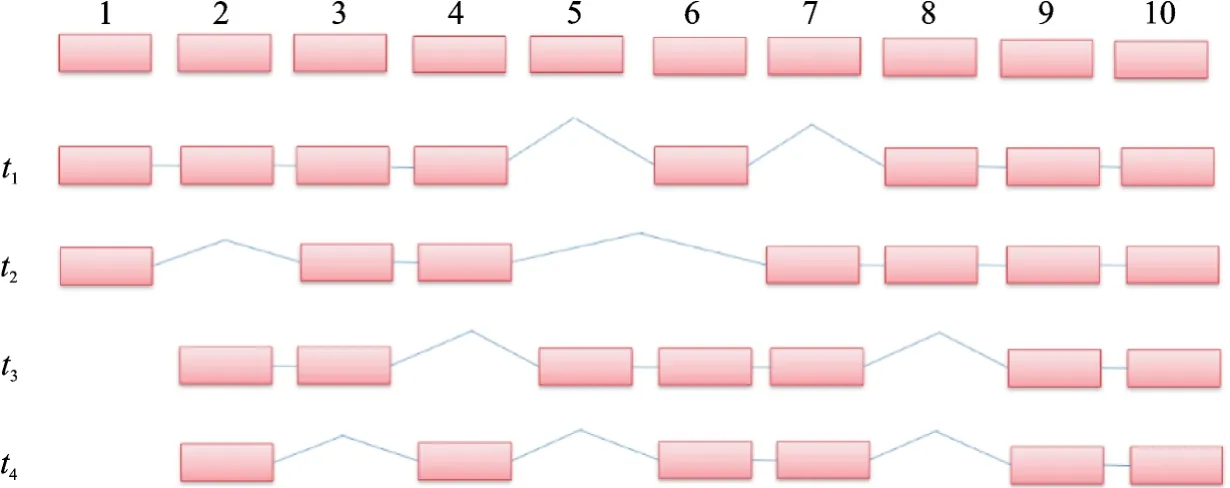
图6 模拟数据结构Fig.6 Structure of simulation data
2.2 非全长读段有效性验证
从表1可以看出当读段长度越长时,非全长读段的数量就越多。因此本文在模拟数据集上做了非全长读段不同占比的对照实验,将100条全长读段按25%,50%和75%的比例随机抽取,并作随机打断,产生相应比例的非全长读段,剩下的全长读段作为对照组,全长读段加上非全长读段作为实验组。在各比例的对照实验中,实验组与对照组均使用DSIDP方法计算结果,并采用计算值与真实值之间的欧式距离作为误差度量。如表2和表3所示,在加入非全长读段数据后,各比例实验的表达水平计算值均比只使用全长读段数据更为精确,表中FL(Full length)表示全长读段,nFL(non-full length)表示非全长读段。值得指出的是,在非全长读段数据占75%的比例时,误差有大幅度的下降,但误差本身仍然要比其他比例只用全长读段数据结果值大,这说明在计算异构体表达水平时,保留非全长读段数据能够降低只使用全长读段数据的计算误差。另外,模拟数据集构建的假设前提是该基因的所有异构体均来自细胞内当前表达且被测序到的mRNA分子,与注释库中的信息无关,因此可以认为当细胞内出现新型异构体时,也能被DSIDP预测出。例如,假设t4为新型异构体,且100条读段数据中包含有t4,则会被DSIDP预测出其结构和表达值。

表2 模拟数据各比例非全长读段计算结果Tab.2 Calculation results on simulation data with different nFL read proportions

表3 模拟数据各比例非全长读段计算误差Tab.3 Calculation error on simulation data with different nFL read proportions
2.3 MCIDP预测异构体验证
MCIDP的提出是为了预测出数据中没有全长读段的超长异构体,本文将模拟数据中t1异构体的所有全长读段随机打断,这时t1异构体即可作为没有全长读段的超长异构体,检验模型的预测能力。实验结果如表4所示,可以看出模型能预测出t1这样的超长异构体,但在表达水平计算上,DSIDP要比MCIDP更精确,原因在于基于马尔科夫链的MCIDP会产生较多低概率的可能路径。如何把这些低概率路径合并至真实异构体中是该类模型后续研究的一个重点。
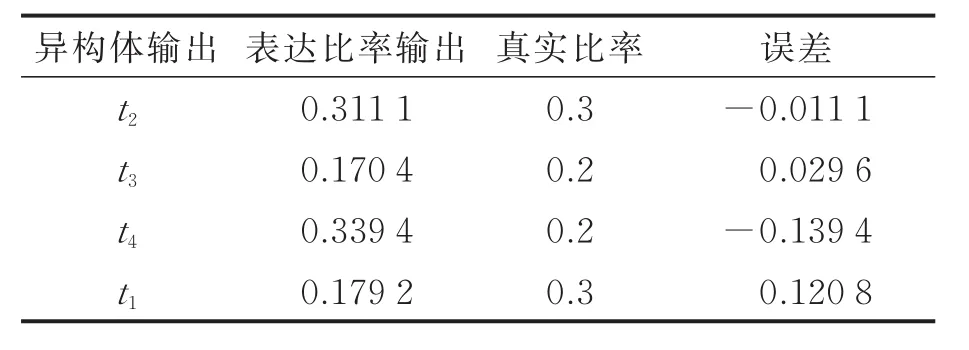
表4 MCIDP在模拟数据上的实验结果Tab.4 MCIDP results on simulation data
2.4 真实数据集实验结果
在异构体表达水平上,虽然ISO-seq数据和RNA-seq数据均反映出样本中原始转录本的浓度,但是由于测序技术本身和数据特性的较大差异,尤其读段长度的差异导致异构体构建上的明显差别,造成两种数据在异构体表达比例计算上的不一致,故无法采用RNA-seq数据的计算结果对ISO-seq分析结果进行验证。因此,对本文中6个cell数据进行分组,分为两次技术性重复实验,具体分组方式和1.3节中的相同。其中Group 1包含139 116个全长读段,147 190个非全长读段;Group 2包含109 969个全长读段,95 431个非全长读段。将本文提出的两个模型应用到这两组重复实验数据中,检验在公共异构体上获得的表达比例的吻合程度,验证本文方法的有效性。
表5给出了两种方法所预测的异构体数量以及注释库异构体数量(注释库为GENCODE数据库中GRCh37-mapped Releases.26),图7展示了表5数据的韦恩图,可以看出MCIDP预测出了更多的异构体,与注释库中已有异构体的交集也更多。因此,MCIDP较适用于注重预测异构体数量的问题中。

表5 模型异构体数量预测结果Tab.5 Number of predicted isoforms

图7 模型预测异构体数量韦恩图Fig.7 Venn diagram of isoform numbers predicted from various methods
另外,注释库是对人类基因的所有已知异构体进行注释,在一些分化后的人体细胞中并不是所有基因都表达,所以图7中注释库中有较多的异构体未被两种模型预测出,而两个模型都预测出了大量不在注释库中的异构体,这在一定程度上也说明了当前注释库还很不完善。图8展示了所提出的两种方法在两次重复实验中计算得到的公共异构体(共4 914个)表达比例的散点图。为了更好地呈现大部分低比例异构体数据的分布情况,本文采用log(105x+1)函数对异构体表达比例进行了变换。经过函数变换,DSIDP结果的相关系数为0.681 7,MCIDP的为0.665 0。可以看出在两组实验数据量不完全一致的情况下,两个模型计算的异构体表达比例也能有较好的一致性,这在一定程度上验证了本文方法的有效性。其中,DSIDP计算的异构体比例在重复实验中的吻合性要高于MCIDP,显示了其更为准确的表达水平和计算能力。

图8 MCF-7数据集异构体层面重复实验结果对比Fig.8 Comparison of repeated experiment results of isoforms in MCF-7 data set
3 结束语
本文在保留ISO-seq数据非全长读段的基础上提出了两个适用于不同场景的异构体预测和表达比例计算模型,DSIDP和MCIDP。两个模型首次仅采用PacBio第三代测序数据用于异构体预测以及表达水平的计算。DSIDP从全长读段中建立异构体预测集合,将所有读段映射至这个集合之中,统计集合元素各自的读段数量,进而计算表达水平,采用Dirichlet采样的方法解决了读段多源映射的问题。实验结果表明DSIDP在异构体表达水平计算上具有较好的准确性。MCIDP是基于马尔科夫链的一个概率模型,通过构造概率图模型,考虑了转录本中所有可能的转录路径,以获得所有可能的异构体。在一些超长异构体无法获得全长读段的情况下,使用MCIDP可以有效地预测出超长异构体。与IDP相比,该模型不依赖异构体的注释信息,只需获取基因的外显子组成即可预测出数据中的异构体,但该模型在计算异构体表达水平上具有一定不足,这与模型存在的低概率相似路径合并这一难点有关。模型中使用的最近距离划分和聚类处理,实际上都是对相似路径的合并,在后续的工作中,拟尝试采用二阶马尔科夫链模型提高相似路径聚类的准确性,以进一步提高异构体比例计算的准确性。
