疾病相关miRNAs识别方法研究进展
2019-05-074
4
(1.广州大学 计算科技研究院, 广东 广州 510006; 2.温州职业技术学院 信息技术系, 浙江 温州 325035;3.广州大学 人事处, 广东广州 510006; 4.广东省数学教育软件工程技术研究中心, 广东 广州 510006)
0 引 言
非编码RNA是一类不能编码蛋白质的RNA分子总称,约占人类基因组总长度的97%,在过去很长一段时间内被称为“垃圾序列”或者“暗物质”.根据序列长度的大小可以将非编码RNA分为3类:<50 nt, 包括microRNA,siRNA,piRNA等等;50~500 nt,包括lncRNA,rRNA,snRNA,snoRNA等等;大于500 nt,包括长的mRNA-like 的非编码RNA,长的不带polyA 尾巴的非编码RNA等等.随着研究的深入,人们发现虽然非编码RNA不能直接编码蛋白质,但是其中一些对于维持细胞内部的平衡,以及与疾病的产生和恶化有着密切的关系.
miRNAs(microRNAs)是一类重要的非编码RNAs,是基因表达和蛋白翻译过程中的调节因子.已有研究表明,miRNA广泛参与细胞的增殖、凋亡及分化生物过程,在肿瘤的发生、形成过程中扮演着十分重要的角色.肿瘤的发生、发展是多条通路联合破坏的结果,因此,肿瘤细胞中的“基因网络特征”比“单分子特征”更能揭示疾病恶化和进展的机制[1-3].由于一个miRNA能够调控多个基因的表达,影响多个信号通路的活性,因此,Chen[4]在著名的新英格兰医学期刊上预测认为,将miRNA当作肿瘤生物治疗的靶分子或者药物将比编码基因更加有效.
权威数据库miRBase收录的miRNAs数目从创办之初的218条,到目前已经增加到38 589条,数量增加了177倍(图1),这一定程度上反映了miRNAs研究领域发展之迅速.此外,笔者通过Web of Science检索关键词“miRNA OR microRNA”梳理了从2001 年至今的发表的相关文献,结果表明,miRNA 相关的研究文章从2001 年8篇到2018年增加至15 587篇,数量增长倍数高达1 900 多倍.最近几年有关miRNA的研究文章,仍处于逐年上升的趋势,这说明miRNA领域的研究依然是当前的热点课题.
目前miRNA研究领域主要集中在以下4个方面:①靶基因的预测.通过开发新的计算方法预测miRNA与基因之间的靶向关系;②miRNA生物学功能作用机制.通过生物学实验的方式研究miRNA在生物体内的功能作用;③疾病相关miRNAs的筛选方法.以miRNA表达谱数据为基础,计算筛选与疾病密切相关的miRNAs;④miRNA小分子药物的开发及应用.以miRNA为药物或者药物靶点,开发适用于治疗特定疾病的新型药物.本文拟从以上几方面阐述疾病相关miRNAs的筛选方法研究进展.
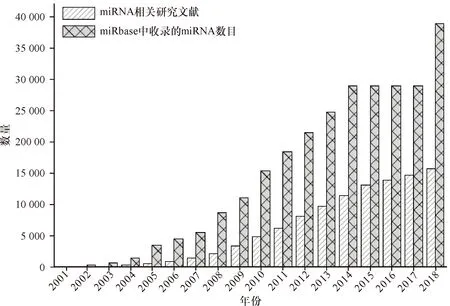
图1 miRNAs相关研究及已发现的miRNAs数量统计数据Fig.1 miRNAs related studies and the statistic data for miRNAs 数据检索时间2019年1月6日
1 miRNA基本概念
1.1 miRNA定义
miRNAs是一类重要的内源性非编码微小RNAs分子,其长度大小约为18~25核苷酸(Nucleotide,nt).在细胞核内,miRNA基因的初始转录产物(pri-miRNA)在RNA聚合酶Ⅲ(RNase Ⅲ)Drosha的作用下切割成为前体miRNA即pre-miRNA[5-7].然后pre-miRNA在转运蛋白(exportin-5)的作用下,从细胞核内转移到细胞质中[8].最后,在另一种RNA聚合酶Ⅲ(RNase Ⅲ)Dicer的作用下,切割生成成熟的单链miRNAs[9].
miRNA在生物体内发挥的功能作用是相当复杂的,总结起来有如下特性:①结合位点差异性.成熟的miRNAs能够通过碱基互补配对的方式结合到靶基因mRNA 3’ 端非转录区域,当完全匹配时候,会促进靶基因的降解,而当非完全匹配时候,则会抑制其靶基因翻译生成对应的蛋白质[10-11];②时空动态特性.当细胞周期阻滞的时候,miRNA能够引导AGO等蛋白与AREs(AU-rich elemens)相结合促进基因的翻译,而在增殖细胞中miRNA对靶基因又起到抑制的作用[12];③位置的影响.除了在细胞质中发挥功能,Xiao等[13]发现miR-24-1在细胞核内通过与基因的增强子区域结合,从而促进基因的表达.
1.2 miRNA命名规则
在研究miRNA之初,miRNA的名称主要是根据其表型进行命名,如lin-4、let-7等等.随着越来越多miRNA的出现,为了方便注释和研究,人们提出了一些关于miRNA的命名标准,目前最新版本的miRBase数据库中关于miRNA的命名准则见图2.

图2 miRNA命名规则简要示意图Fig.2 A brief sketch of the naming rules for miRNA.
miRNA成熟体命名规则如下(以动物miRNA为例):
(1)在统一命名规则制定之前发现的miRNA,则继续保留原来名字,如hsa-lin-4;
(2)miRNA的成熟体表示为miR,然后根据其物种类型,以及被发现的先后顺序标注阿拉伯数字,如hsa-miR-21;
(3)若多个miRNAs高度同源,则在数字后面加上英文小写字母(a,b,c,…),如hsa-miR-34a,hsa-miR-34b等;
(4)由不同染色体上的DNA序列转录加工而成的具有相同成熟体序列的miRNAs,就在后面添加上阿拉伯数字,如hsa-miR-199a-1和hsa-miR-199a-2;
(5)通常一个miRNA前体长度大约为70~80nt,有时候2个臂分别会产生成熟体miRNAs.在之前人们的做法是:表达量比较高的miRNA后面不添加任何符号,而对于表达量比较低的miRNA后面则加上*号,如rno-miR-9*.而最新版本的miRBase数据库中则以“-5p”和“-3p”对成熟miRNAs分别命名.如hsa-miR-21-5p和hsa-miR-21-3p,分别表示从前体has-mir-21的5’端臂和3’端臂产生而来.
2 miRNA靶基因预测方法
已有研究表明,人类相关基因中大约有三分之一的基因会受到miRNA的靶向调控.到目前为止,虽然陆续有生物学实验证实了一些miRNAs与mRNAs之间的靶向调节关系,但是仍然还有许多靶基因没有经过生物学实验验证,这一定程度上阻碍了miRNAs生物学的功能被充分的发掘[10, 14-15].因此,关于miRNAs一项重要的研究内容同时也是极具挑战性的工作,就是如何快速、准确地预测以及验证miRNAs的靶基因.该项研究对于探究miRNAs生物学功能作用、参与涉及的生物信号通路以及它在疾病的发展过程中扮演的角色具有重要的意义,与此同时,也能够加快miRNAs作为一种治疗靶点或者药物走向临床实践.
借助计算生物学的专业优势,将其用来替代生物学家大量的分子克隆实验工作,基于这种研究模式获得了非常好的效果[16-18].但是,预测动物的miRNAs靶基因是一项极具挑战性的工作,其主要原因是由于许多动物的miRNAs与其靶基因mRNAs结合位点并非完全互补的.除此以外,由于已知的经过实验验证的靶向关系比较少,所以在评价预测结果的时候,没有足够的金标准来作为客观的参考对象.到目前为止,对于计算机方法预测、推断出来的靶基因,还没有一个快速、可靠的高通量验证鉴定方案,这在一定程度上影响了算法准确性的评价,也阻碍了计算预测算法的更进一步的优化及改进[19].从成为生命科学界的研究热点到目前为止,miRNAs相关生物学功能的探索发现研究工作已经历了10多年的发展历程,关于miRNAs靶基因的预测方法已经发展出数十种之多.这些靶基因预测计算方法主要可以划分为2大类:ab initio计算方法和机器学习预测方法.总体上来说,刚开始提出的计算预测方法均属于ab initio计算方法,这些预测方法都是基于实验获得的结构特征来引导预测方法的开发.机器学习预测方法主要是基于实验训练集的相似度来预测潜在的靶向目标.
2.1 ab initio方法
(1)miRanda[20-21]:MiRanda以及相应的网站microRNA.org是由美国著名的癌症研究中心——斯隆-凯特琳研究所(Sloan-Kettering Institute,SKI)开发的在线查询数据库,该数据资源可以非常方便地免费在线获取.miRanda的最新版本发布时间是在2010年8月,该最新版本也称为mirSVR.该方法适用范围广,不受物种限制.MiRanda采用Smith-Whatman算法的思想[22],利用该方法来检测互补匹配的序列(允许位于miRNA的5p端第2-7nt种子序列有一个G∶U匹配或者错误匹配的存在),除此以外,这一算法还增加了关于RNA二级结构预测的程序,从而可以从动力学的角度去考察miRNA与其候选靶基因之间的结合稳定性;根据前面的准则得到了靶基因结合位点,然后考察这些结合位点在不同物种间的保守性,最后给出自由能(△G)以及序列匹配分数(Score).miRanda靶基因预测方法主要考虑了以下几个方面因素:①靶向结合序列的互补性;②mRNA序列的二级结构对miRNA-mRNA结合稳定性的影响;③靶基因结合位点在跨物种间的保守性.
(2)TargetScan[23-24]:是由Lewis等于2003年开发出来的miRNA靶基因预测工具,该方法首次引入假阳性率来评估预测结果.TargetScan靶基因预测算法主要考虑如下2个方面特点:①考察miRNA靶向基因mRNA的3p端非转录区序列在跨基因跨物种间的保守性;②研究miRNA与靶基因mRNA形成的miRNA-mRNA双链结构的动力学稳定性[25].该方法要求miRNA的5p端的第2-8位碱基(种子序列)与靶基因mRNA的3p端必须满足互补匹配,然后要求种子序列完全互补的情况下从种子序列向两端扩展,直到遇到不能配对的碱基为止,这个过程中允许G-U配对.该方法通过计算靶基因结合位点产生的自由能以及信噪比来评价计算机预测结果的准确性.
(3)PicTar[26]:该算法同样要求miRNA 5P端1-7nt或者2-8nt种子序列在靶基因位点识别中的关键因素,主要强调miRNA与其靶基因形成的二聚体结合能在靶基因翻译抑制过程中的影响.PicTar将种子序列分为2类:“完全匹配的种子序列”和“不完全匹配的种子序列”,前者需要种子序列与靶基因序列完全匹配,后者则可以在满足miRNA靶基因二聚体结合能要求的情况下允许种子序列出现错配(不允许G-U配对).
(4)DIANA-microT:基于实验和计算预测方法,Kiriakidou等于2004年开发出一种“DIANA-microT”miRNA靶基因预测程序[27].该方法结合了实验与计算机计算方法来预测哺乳动物的靶基因,这个方法的一大特点是它着重考虑了单一结合位点(miRNA∶mRNA)的miRNA靶基因.在筛选miRNA靶基因的过程中,DIANA-microT不仅考虑了重要的种子序列区域,而且考虑了miRNA的3p端与靶基因mRNA的结合.在识别靶基因时候,不仅考虑保守位点,而且也考察了非保守结合位点.
(5)PITA[28]:该算法由Kertesz等于2007年提出,考虑了特定二聚体互补匹配信息,并且引入了miRNA位点可接近性的概念.miRNA位点可接近性表示整个二聚体的最小自由能与互补匹配区域的原始能量之间的差值.用户可以通过调整约束条件来选择候选靶基因列表(种子序列最小长度,G∶U错配与未配对个数).
2.2 机器学习预测方法
(1)TargetBoost[29]:该算法采用GPboost模型,基于miRNA与候选靶基因mRNA形成的miRNA-mRNA二聚体的序列互补匹配程度、热动力学稳定性、以及跨物种保守性等特征,来推测线虫和果蝇的miRNA的靶基因.该算法使用的正样本为36个实验验证的miRNA-mRNA靶向关系数据,而用于训练的负样本数据集为300个随机生成长度为30 nt的基因序列.
(2)miTarget[30]:该算法使用支持向量机方法,以及径向基函数,预测miRNA靶基因.该方法考虑了miRNA-mRNA二聚体的结构特征、热动力学性质和碱基互补匹配程度等特征.miTarget算法没有考虑靶基因在多种物种间的序列保守性.miTarget算法中用于训练支持向量机的负样本数据集,涉及83个经过实验验证的miRNA-mRNA靶向关系,以及163个通过实验数据推理得到的miRNA-mRNA靶向关系数据,正样本数据集包含152个miRNA-mRNA靶向关系数据.
(3)MiRTif[31]:该方法首先整合miRanda、PicTar以及TargetScan这3种miRNA靶基因预测方法得到的各种特征得分,然后使用支持向量机方法预测miRNA候选靶基因,核函数采用径向基函数.MiRTif方法使用到的正样本数据集由195个实验验证的miRNA-mRNA靶向关系数据构成,负样本数据集囊括21个生物学实验验证的和17个假定的miRNA-mRNA靶向关系数据.
(4)MTar[32]:该算法重点考虑了3类区域的靶基因结合位点:仅仅考虑5’端种子区域;以5’端种子区域为主;以及以3’端种子区域为主.首先计算相应区域miRNA-mRNA双链的特征得分,然后采用人工神经网络方法计算预测miRNA靶向基因.Mtar算法使用了340个miRNA-mRNA靶基因数据作为正样本数据集,随机产生了400个miRNA-mRNA靶基因数据作为负样本数据集.
(5)miRTDL[33]:该算法主要考虑了miRNA-mRNA之间的碱基互补匹配性、可接近性以及序列的保守性等特征.本方法选择了1 297个实验验证的miRNA靶基因数据作为正样本,309个实验验证负例样本.因为该方法中负样本远少于正样本的数量,所以该方法首先利用约束松弛方法构建了均衡的正、负样本数据集,然后再利用深度学习模型,推测miRNA的靶基因.
miRNA从1993年被首次发现,到成为研究热点,持续至今已经历20多年的历史,在这个过程中,围绕miRNA,科研学者积累了大量的数据资源,并建立了数据库供同行分享使用,以便人们能够在miRNA研究道路上走的更好、更远.关于miRNA的数据库系统已有数十种甚至多达百种,本文总结了一些常用的数据库(表1).这些数据库中有些是关于miRNA功能注释的(miRbase,miRDB),有些是有关miRNA靶基因信息预测的数据库(miRanda,TargetScan,PITA等).
通过回顾总结miRNA靶基因计算预测的工作,可以发现这些方法都有如下一些特点:miRNA与其靶向mRNA序列跨物种、跨基因间的保守性;miRNA的5p端在miRNA-mRNA互补匹配中的重要作用;miRNA与靶基因mRNA形成的双链的二级结构以及能量特征.不同的预测方法有其一定的使用范围和独特优点,相信随着实验验证方法的不断发展,各种计算预测算法也可以相互补充完善,从而提高靶基因预测的精确度.
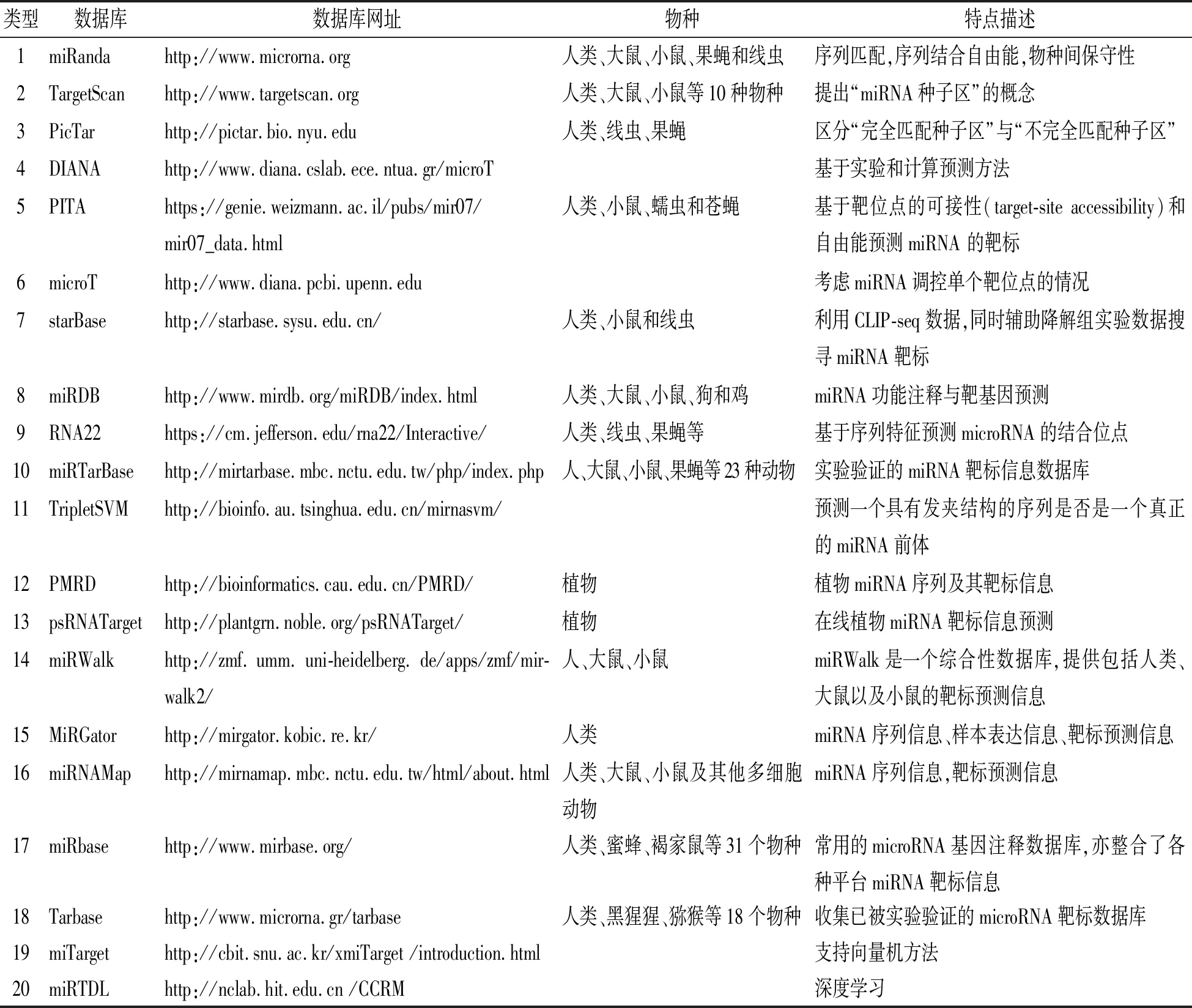
表1 miRNA靶基因预测数据库
3 miRNA调控机制
Lee等[34]于1993年在做筛选线虫时序相关基因的研究时,发现了一个长度仅有22nt的RNA小分子——lin-4,这个小分子不编码蛋白质,而是以碱基互补配对的方式结合到lin-14基因的3’端非转录区域(3’UTR)的特定位点,抑制lin-14对应mRNA的翻译,最终使得其蛋白质合成的量明显降低,因此,称其为转录后抑制,这是首次发现miRNA具有调控基因表达功能的研究.当时人们并不能完全理解为何如此微小的RNA分子会具有调控基因表达的功能,随后关于该领域的研究一直沉寂了7年之久才再次有新的发现.2000年,Reinhart等[35]同样是在研究线虫C.elegan时序数据时,发现了另一个长度只有21 nt小分子RNA——let-7,经过研究发现,该小分子能够靶向基因lin-41,从而导致该基因对应的蛋白质表达量减少.后来,随着不断的深入研究,生物学家总结出miRNA的功能作用机制,即成熟的miRNA能够通过碱基互补配对的方式结合到靶基因mRNA上,当种子序列完全互补匹配时会促进靶基因的降解,而当不完全互补匹配时则会抑制靶基因的翻译生成对应的蛋白质.
长期以来,人们一直认为miRNA对于靶基因的表达是起到负调控的作用,因此,在研究miRNA功能的时候会选择性地忽略正相关的miRNA-Gene调控关系,仅仅考虑具有负相关的miRNA-Gene调控关系.然而也有一些打破传统约束,揭示出令人惊喜的研究成果,Vasudevan实验室2007年发表在Science杂志上的一篇文章,发现miRNA并非总是负调控基因的表达,在一些特定情况下miRNA能够起到促进基因表达的作用.该课题组通过实验发现,miR369-3能够引导AGO等蛋白与AREs(AU-rich elemens)相结合促进基因的翻译,除此以外,该课题组还发现在静态细胞中即G0期状态下let-7能够与人工合成的miRcxcr4协同促进靶基因的翻译表达,而在增殖细胞中它们对靶基因又起到抑制作用[12].2017年复旦大学于文强课题组在RNA Biology上发表了一篇文章,表明miRNA在细胞核内可以与增强子结合,从而改变增强子的染色质状态,进一步达到激活基因的转录表达的结果[13].曾获得诺贝尔生理学奖的Phillip Sharp同样于2017年在Cell上提出了miRNA激活理论[36],文章指出miRNA在细胞核内则会与增强子相互作用,这也一定程度上肯定了复旦大学于文强课题组的研究成果.
4 疾病相关miRNAs筛选方法
已有研究表明,包括肿瘤在内的许多复杂疾病的发生、发展过程中往往伴随着编码基因以及非编码基因尤其是miRNA的差异化表达,如何识别在疾病进展过程中异常表达的miRNAs,对于疾病的诊断以及新药的开发具有十分重要的意义.生物学家通过实验的方法发现了一些疾病相关的miRNAs,哈尔滨工业大学蒋庆华课题组[37],北京大学崔庆华课题组[38],以及Yang等[39]基于文献搜索方式,人工整理了与疾病相关的miRNAs(表2).除了通过实验的方法以外,近年来关于筛选疾病相关miRNAs,出现了多种基于计算的方法,本节主要分3个部分来叙述:基于miRNA表达浓度的分析方法;基于miRNA表达相对位置排序的分析方法;基于miRNA功能相似性网络分析方法.

表2 人类疾病相关的miRNAs数据库
(1)基于miRNA表达差异分析方法
直接法:直接法的意思是直接基于miRNA表达水平做差异表达分析,该方法假设如果某个miRNA差异表达,那么它对应的下游靶基因就是差异表达的.分析miRNAs表达谱最简单常用的方法是倍数法(FC,Fold Change)以及T-test差异检验方法[40-41].倍数法是基于miRNAs表达浓度计算变化的倍数,选取变化大的miRNAs作为候选的疾病相关miRNAs.倍数方法操作简单方便,同时它也有显而易见的缺陷,就是这种方法缺少差异显著性估计值,使得倍数阈值的选择比较随意,会受到主观意愿的影响(到底是1.5倍、2倍或者还是3倍?).T-test差异检验方法可以弥补倍数法的不足,该方法可以通过设置显著性阈值来获得表达差异比较显著的miRNAs.T-test差异检验方法是基于miRNAs的表达浓度呈正态分布假设为前提的.不管是倍数法还是T-test差异检验方法,他们都会受到实验批次效应(Batch effect)和数据标准化过程的影响[42].
间接法:miRNA与靶基因mRNA之间是一种“多对多”靶向调控关系,因此,简单地通过单个miRNA的表达变化情况来决定其靶基因是否差异变化存在明显的不足之处,因此,一些综合的分析方法被提出,即通过多个miRNAs的协同作用间接确定下游靶基因的差异变化.东南大学肖忠党课题组于2016年提出了基于miRNA表达浓度以及miRNA-mRNA结合系数,综合评估miRNA表达谱整体对下游靶基因的影响[43],为系统性筛选受miRNA表达谱全局性显著调控的编码基因提供了解决方案.Garcia-Garcia等[44]提出基于多个miRNAs表达变化叠加效应来确定下游靶基因是否差异表达,该模型中考虑了miRNA水平差异表达变化的方向(+/-)以及显著性值P-value.
(2)基于miRNA表达量的排序方法
哈尔滨医科大学顾云燕课题组等认为在个性化样本内,一些miRNAs在正常样本中是存在着相对稳定排序的,而疾病状态下有些miRNAs会发生排序的逆转,基于该设想,顾云燕课题组提出基于miRNA表达相对位置排序筛选差异变化的miRNAs.该课题组首先在正常样本群体内筛选具有稳定表达顺序的miRNAs基因对,然后检验个性化样体中miRNA排序的差异变化来筛选差异表达的miRNAs[45].在编码基因水平,已有研究指出,基于基因排序筛选差异表达基因的方法可以大大地降低对于实验批次效应以及数据标准化过程带来的负面影响[46-47].虽然该方法对于抵抗批次效应以及数据标准化过程带来的负面影响,是个非常有效的方法,然而它却是牺牲了表达谱中关于miRNA的精确的量化信息.
(3)基于miRNA功能相似性网络方法
单个miRNA能够靶向调控多个mRNAs的表达翻译,这些mRNAs可能富集在某些GO功能模块中.如果2个miRNAs能够协同调控某一个公共的GO功能模块,那么这2个miRNAs就被认为具有功能协同的作用.基于该思想,哈尔滨医科大学李霞课题组等构建了miRNA-miRNA协同调控网络,从miRNAs协同调控网络的角度探究肿瘤疾病相关的miRNAs,为开发miRNAs药物或者发现新的治疗靶点提供了一个新的视角[48].Li等[49]采用多个miRNAs协同调控靶基因mRNAs的思想,基于miRNA、mRNA表达谱数据提出了一种构建miRNA-mRNA调控网络的分析方法.功能相似的miRNAs 调控的疾病也应该比较相似,基于这样的假设,郭茂祖等[50]提出了BNPDCMDA算法.该方法首先构建了miRNA-疾病双层网络;然后,通过miRNAs 的功能相似度对其进行基于密度的聚类;最终,将聚类后的miRNAs与疾病构成的miRNA-疾病双层网络,采用二分网络投影方法预测miRNA-疾病关联.
除了以上3类从miRNAs表达谱数据出发逐步筛选疾病相关的miRNAs以外,也有一些从编码基因表达谱数据出发的逆向反推导的方法.赵兴明课题组仅利用编码基因表达谱数据,提出了筛选疾病相关miRNAs的研究方法[51].本课题组在前期的工作中提出基于基因子通路的方法筛选疾病相关miRNAs,该方法用到的数据是编码基因表达谱数据,首先对基因数据集做差异表达分析,然后对差异表达的基因做子通路富集分析,最后根据miRNA-mRNA作用关系筛选疾病相关的miRNAs[52].
5 miRNA与小分子药物
已有研究表明,生物活性小分子或者药物能够调控miRNA的表达,这预示通过小分子药物靶向调控miRNA或许能够为疾病带来新的治疗方案.2008年Gumireddy实验室开发了首个靶向miRNA的小分子抑制剂,该抑制剂被设计主要用来靶向抑制miR-21的表达[53].自此以后,人们利用高通量筛选或者通过计算机序列比对方法设计出大量的针对非编码miRNA的小分子抑制剂.2012年哈尔滨医科大学李霞课题组基于miRNA表达数据以及药物对转录组的影响结果,提出一种方法,将miRNA与小分子药物联系起来,构建了一个网络SMirN[54],为发现新的治疗靶点或者治疗药物提供了辅助性工作.2013年哈尔滨医科大学李霞课题组建立了一个miRNA与小分子药物关系数据库SM2miR,专门收集实验验证的能够靶向调控miRNA表达的小分子药物[55],该数据库包含了2 925个作用关系(涉及151个小分子药物,747个miRNAs以及17个物种).2017年上海同济大学赵兴明课题组构建了一个miRNA影响药物治疗效果的关系数据库[56],该数据库不仅存储了miRNA与小分子药物的对应关系,而且还给出了miRNA对药物治疗效果的影响.
随着miRNA相关研究的不断深入,一些针对包括肿瘤在内的多种疾病的miRNAs药物被开发出来,并且陆续有一些miRNA药物从实验室走向临床试验阶段(表3).miRNA小分子药物相关数据库的构建必将有效地加速推进miRNA在临床医学中的具体应用.
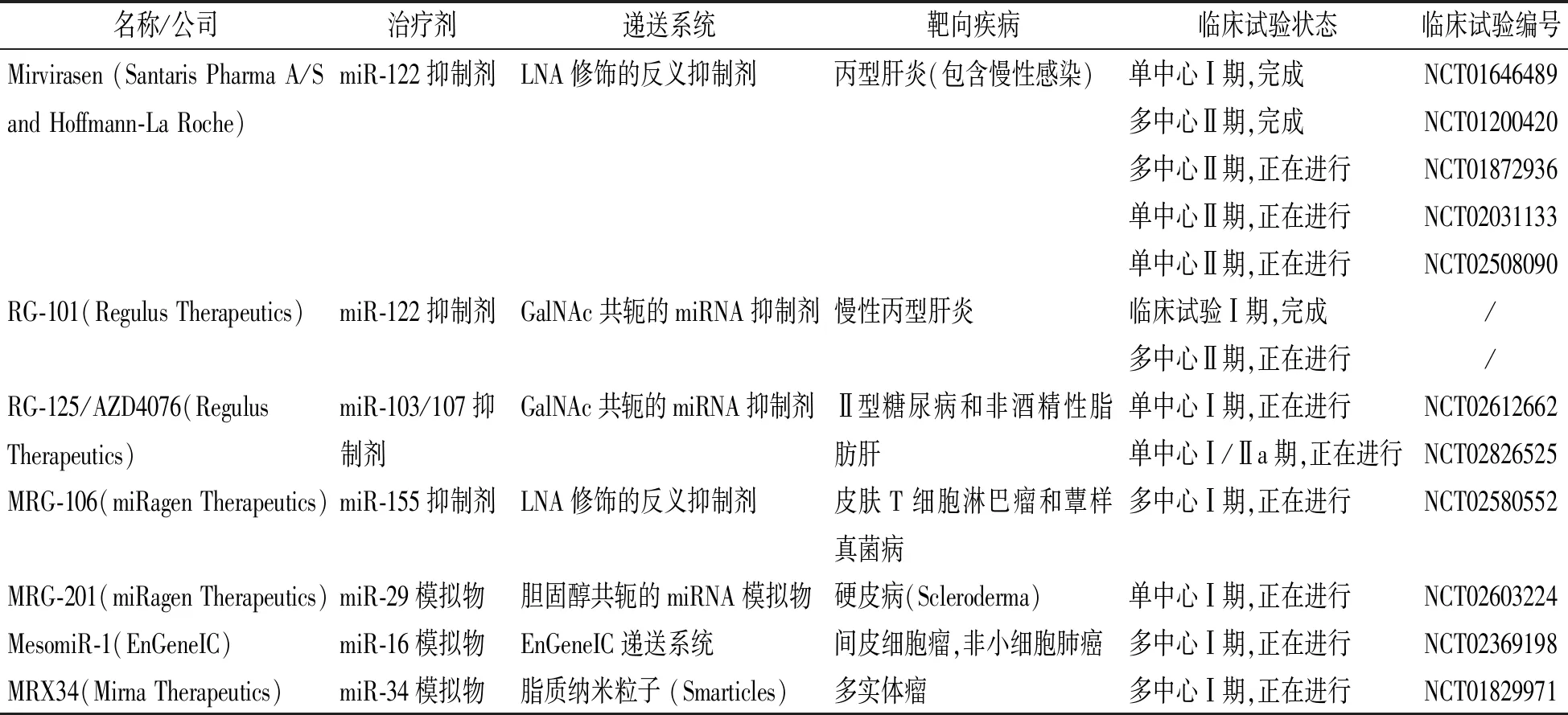
表3 进入临床试验中的miRNAs药物[57]
6 总 结
随着miRNA研究的不断深入以及高通量测序技术的发展,人们在靶基因预测、miRNA生物学功能机制的探究以及疾病相关miRNAs筛选等方面,发展了大量的分析研究方法,并发现了许多对疾病具有重要影响的miRNAs,其中一些对疾病治疗极具潜力的miRNAs药物已经进入临床试验阶段.
虽然关于miRNA的研究已有十几年的历史,并且取得了喜人的丰硕成果,但是还有一些方面有待继续跟进:①靶基因预测方法普遍存在高假阳性的缺点,靶基因预测准确性对于miRNA功能的研究至关重要,因此,亟待发展具有突破性的计算方法来预测miRNA的靶向基因;②miRNA对靶基因调控的复杂性,miRNA与靶基因之间是一种多对多的靶向关系,且miRNA对靶基因具有抑制或者促进的双重作用,因此,如何准确量化miRNA对其靶基因的影响是一个具有挑战性的重要课题;③加速推进miRNA药物走向临床,筛选疾病相关的miRNAs,其重要目的在于将其推向临床医学实际应用,可喜的是已有些miRNAs药物进入临床试验阶段,遗憾的是目前还没有一个合格的miRNA药物被批准进入市场.因此,有待新的有效筛选方法被提出,从而加速miRNA药物早日进入临床治病救人.
