同基因型同卵双胞胎姐妹同患血卟啉病的不同临床表现
2019-04-11王晓洁赵莘瑜焦淑洁王天舒刘晓宇
王晓洁, 赵莘瑜, 焦淑洁, 王天舒, 孙 琦, 刘晓宇
血卟啉病(Porphyria)是由于血红素合成途径中有关酶的缺乏导致卟啉类化合物代谢紊乱而发生的一种罕见疾病,以腹痛、神经精神症状及光感性皮肤损害为主要特征。血卟啉病高发于20~40岁女性,患病率(0.5~10)/10万[1],临床上以急性间歇性血卟啉病(acute intermittent porphyria,AIP)最为常见。本病患病率低,临床表现各异,误诊率高,目前对该病的认识与研究仍较局限。查阅文献尚无同卵双胞胎同患血卟啉病的相关报道。现就我院收治的一对同卵双胞胎姐妹同患AIP的相关临床资料分析如下。
1 临床资料
1.1 例1(姐姐) 女,24岁,以“腹痛14 d,抽搐4 d”于2017年12月9日入我院神经重症病区。14 d前不明诱因出现发热、腹痛,热峰达37.8 ℃,停止排便,伴纳差、进食少,偶伴恶心、呕吐,呕吐物为胃内容物。1 w前服用食用油排便后症状稍缓解。4 d前无诱因出现发作性意识丧失,四肢抽搐、头后仰、双眼上翻、牙关紧闭,伴小便失禁,持续4~5 min缓解,当天共发作3次,无头痛、肢体无力、精神行为异常。就诊于当地医院,脑MRI发现双侧额顶颞叶异常信号,腹部CT考虑不全肠梗阻,给予禁食及其他治疗(不详),近1 d腹痛加重,就诊于我院。查体:血压175/125 mmHg,心率120次/min,律齐,各瓣膜听诊区未闻及杂音。腹部平坦,腹肌轻度紧张,右侧腹部压痛明显,肝脾未触及,肝区叩击痛阳性,肠鸣音可闻及。神经系统查体无异常。
入院后完善相关检查:血常规:中性粒细胞80.7%↑;淋巴细胞11.0%;中性粒细胞6.86×109/L↑;电解质:钾3.17 mmol/L↓;钠132 mmol/L↓。尿常规:比重1.005↓。肝功能:谷丙转氨酶85 U/L↑;谷草转氨酶88U/L↑。心肌酶:肌酸激酶1600.0 U/L↑;CK同工酶MB19.00 U/L↑;N端脑利钠肽309.08 pg/ml↑。血脂:总胆固醇5.80 mmol/L↑。同型半胱氨酸37.35 μmol/L↑。抗甲状腺过氧化物酶抗体174.70 IU/ml↑;抗甲状腺球蛋白抗体264.00 IU/ml↑。脑脊液常规、生化、病毒、细菌、结核、寄生虫、电泳检查无异常。肾功能、结缔组织全套无异常。性激素:睾酮1.00 ng/ml↑。皮质醇节律:皮质醇上午8点<1.00 μg/dl↓。胸部、全腹部增强CT:双下肺慢性炎症,未见肠梗阻。彩超:甲状腺左侧叶低回声结节(TI-RADS分级:3级)甲状腺左侧叶囊实性结节;心脏、肝胆胰脾、双肾、颈动脉、双下肢深静脉、腹主动脉彩超无异常。头部MRI平扫+增强:双侧额顶颞枕叶皮质及皮质下见片状脑回样长T1长T2异常信号影,FLAIR呈高信号,DWI高b值呈稍高信号,双侧额顶颞枕叶异常信号未见明确强化。给予抗癫痫、营养神经、改善循环、抗感染、清除自由基、维持电解质平衡等对症治疗。
入院2 d查体四肢肌力4级,四肢腱反射消失。3 d四肢肌力3级,腱反射消失。患者症状进行加重,间断腹痛、癫痫发作、自主神经功能紊乱、肌力减弱、腱反射消失,结合患者病史、症状、体征及相关检查,考虑患者血卟啉病可能性大。入院5 d(家属强烈要求转至我科),精神差,反应迟钝,计算力差,双侧软腭上抬无力、咽反射减退,四肢肌张力低,双上肢远端肌力约3级,近端0级,双下肢肌力1级。四肢腱反射未引出,病理征未引出,脑膜刺激征阴性。尿液实验:尿液日光下暴晒后由洗肉水色变为深红色(见图1)。考虑血卟啉病,给予高糖饮食,静脉滴注10%葡萄糖、营养神经、补充营养等对症治疗。入院12 d,双上肢远端肌力降至2级。加用激素治疗,症状缓解仍不明显。入院14 d出现呼吸表浅,三凹征阳性,咳嗽反射明显减弱。双上肢肌力1级,双下肢踝关节上7 cm以下、双上肢腕关节以上5 cm以下痛觉过敏。病变累及呼吸肌,有呼吸衰竭风险,再次转入神经重症必要时气管切开。入院20 d复查头部MRI:双侧顶叶见斑点状等T1等T2信号,黑水像呈等信号,DWI呈高信号。病变呈现可逆性改变(见图3~图5)。患者家属拒绝继续治疗出院。
1.2 例2(妹妹) 女,24岁,以“间断腹痛5 y,再发并加重3 d”为主诉于2017年12月18日入院。5 y前进食后出现脐周疼痛,持续隐痛,可耐受,无恶心呕吐,无发热、腹泻。后上述症状间断发作,1 y前来我院查腹部平片:肠胀气,以“腹痛原因待查”住消化科,查腹部CT:盲肠、升结肠、横结肠明显扩张积液、积气。腹部彩超、腹主动脉、肠系膜动、静脉无异常。头部MRI:左侧脑室三角区旁可见小片状长T1长T2信号,黑水像呈高信号,DWI高b值未见弥散受限(见图6)。给予禁食、灌肠、抗感染等治疗后好转出院。5 y来间断腹痛,常为脐周持续隐痛,自服药物效差,持续3 w可自行缓解。3 d前无诱因再次出现腹痛,较前加重。查体无异常。入院后查:血常规:白细胞5.80×109/L;红细胞3.79×1012/L↓;血红蛋白102.0 g/L↓;血小板112×109/L↓;红细胞压积0.302 L/L↓;平均红细胞体积79.50 fl↓;平均红细胞血红蛋白含量26.80 pg↓。肝功能:前白蛋白102 mg/L↓。肾功能:尿素8.30 mmol/L↑;肌酐130 μmol/L↑;尿酸970 μmol/L↑。甲状腺功能:T3:5.99 pmol/L;T4:33.83 pmol/L↑;TSH:0.01 μIU/ml↓。同型半胱氨酸39.70 μmol/L↑;叶酸、维生素B12、糖化血红蛋白、电解质、传染病检查无异常。心电图:窦性心动过速。尿液实验:太阳暴晒下呈葡萄色(见图2)。给予10%葡萄糖、降同型半胱氨酸等治疗后,腹痛症状明显缓解出院。
应用高通量测序方法,分析例1卟啉病相关致病基因HMBS、PPOX、HFE、ALAD、FECH、ALAS2、UROD、UROS、CPOX基因各外显子编码区及剪接区的变异情况;应用PCR结合Sanger测序技术分析例1、例2及患者父亲HMBS基因c. 518G>A(p. Arg173Gln)位点的变异情况。结果:例1、例2及患者父亲均存在HMBS基因c. 518G>A(p. Arg173Gln)杂合变异(见图7)。
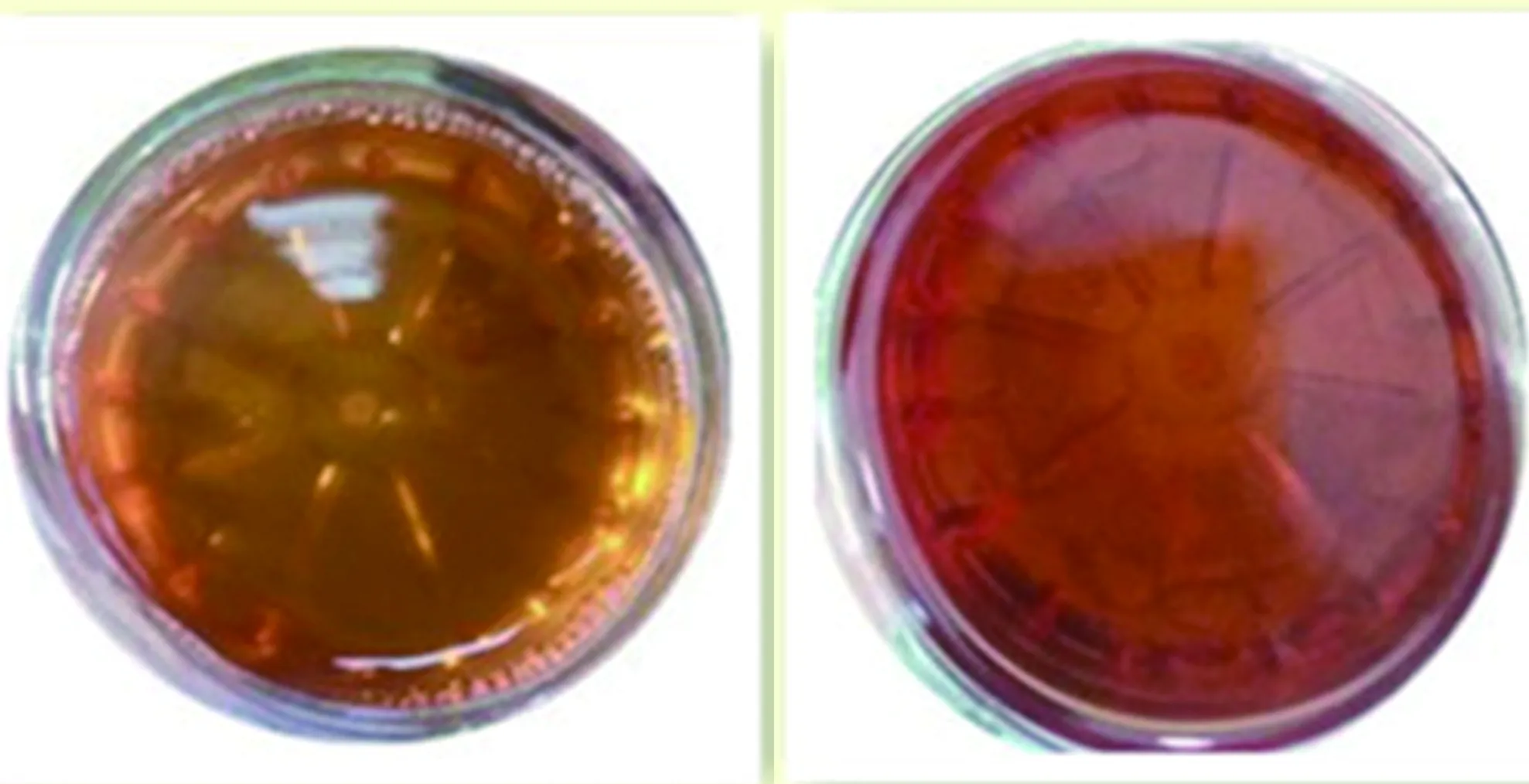
图1 例1尿液阳光照晒前后对比(左图为晒前,右图为晒后)

图2 例2尿液阳光照晒前后对比(左图为晒前,右图为晒后)

图3 例1MRI T1、T2、Flair序列前后对比(第一排为治疗前:广泛额顶颞枕叶病变。第二排为治疗后:顶叶斑点状病变)

图4 例1 MRI DWI序列前后对比(第一排为治疗前:广泛额顶颞枕叶病变。第二排为治疗后:顶叶斑点状病变)

图5 例1第一次磁共振增强图(广泛额顶颞枕叶病变)
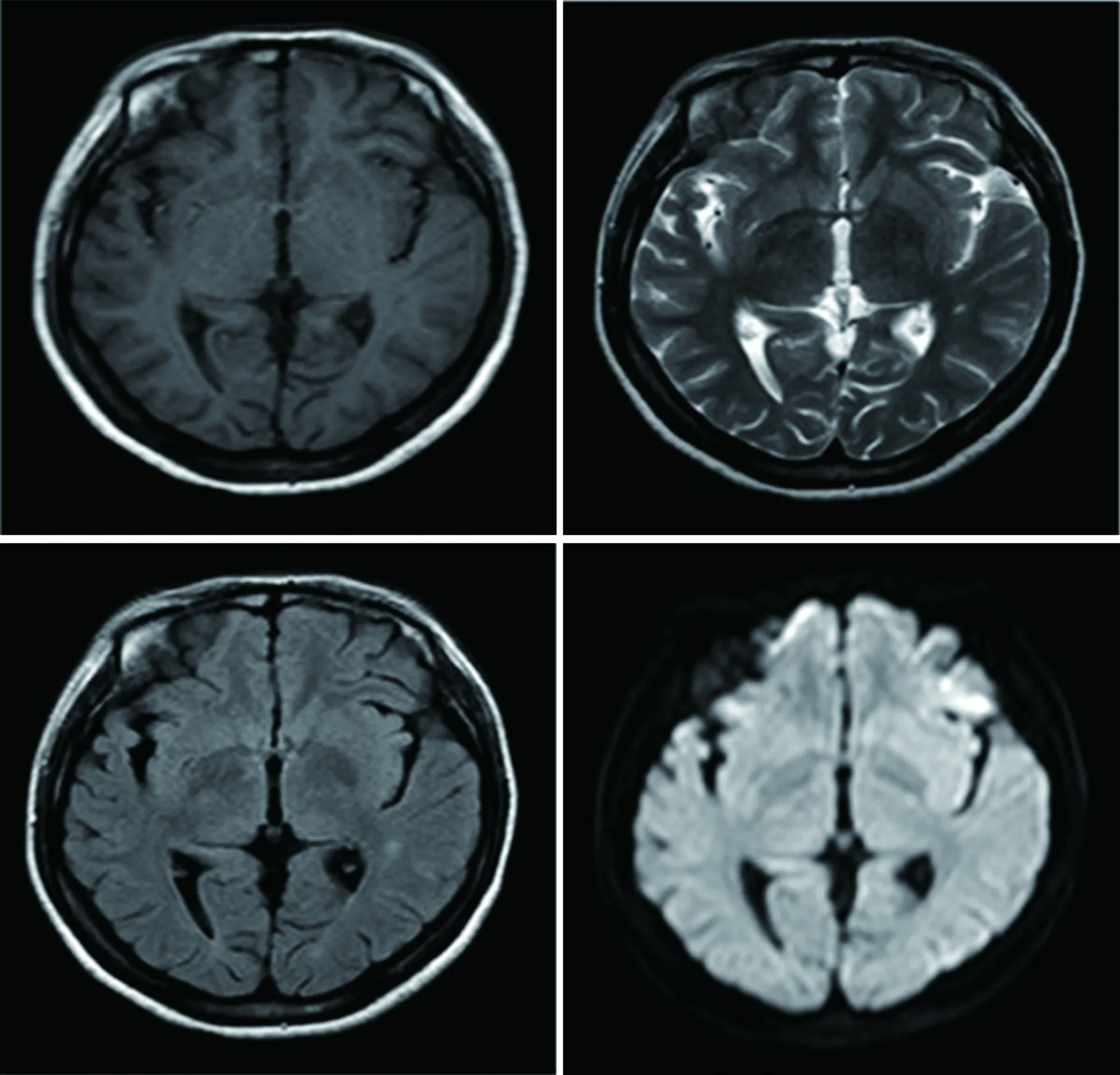
图6 例2头部MRI左侧脑室三角区旁小片状缺血灶

图7 患者父亲、例1、例2基因图 (上图为患者父亲,中图为例1,下图为例2) 同为c. 518G>A(p. Arg173Gln)杂合变异
2 讨 论
本组2例为双胞胎姐妹,两人长相、身材极为相似,据其家属提供信息两人为同卵双胞胎。例1于24岁发病,以腹痛为首发症状,后迅速累及中枢神经系统、周围神经系统并自主神经紊乱。查头部MRI示双侧额顶颞枕叶皮质及皮质下异常信号影。给予对症及高糖治疗后,症状改善不明显,逐渐累及呼吸肌,呼吸表浅、呼吸费力。20 d后复查头部MRI:双侧顶叶点状弥散受限。例2于19岁起出现间断腹痛,病程5 y,未累及中枢、周围神经系统,无精神症状。头部MRI:左侧脑室三角区异常信号,考虑缺血灶。给予高糖治疗后,腹痛明显缓解。二者尿液暴晒后均成酒红色。因条件限制,二者均未应用血红素治疗。患者母亲因病去世(不详),其父女三人行DNA检查,三者均为同一基因型。综上所述,尽管同卵双胞胎姐妹同患AIP,其临床、影像学表现及预后完全不同。推测原因:(1)疾病严重程度除与基因型有关外,还受环境及其他因素影响。(2)发病累及中枢神经系统,影像学表现明显者病情较重,尽管影像学表现可逆转,但高糖等相关治疗不能逆转已有损害。
血卟啉病(Porphyria)是因血红素合成路径中有关酶的缺乏导致卟啉类化合物代谢紊乱而发生的疾病,依据血红素前体物质(ALA、PBG 以及卟啉类化合物)异常合成或蓄积的主要组织部位,将卟啉病分为肝性血卟啉病和红细胞生成性血卟啉病。肝性血卟啉病根据临床表现的急剧程度又分急性和慢性两类。其中急性肝性血卟啉病包括急性间歇性卟啉病(AIP)和遗传性粪卟啉病(HCP)、混合型血卟啉病(VP)及ALA 脱水酶缺乏型血卟啉病(ALADP)。慢性肝性血卟啉病包括PCT和肝红细胞生成性卟啉病(HEP)。红细胞生成性卟啉病可分为先天性红细胞生成性血卟啉病(CEP)和红细胞生成性原卟啉病(EPP )。肝细胞线粒体的ALA合成酶(ALAS)催化甘氨酸和琥珀酸辅酶A 合成5-氨基乙酰丙酸(ALA),ALA从线粒体内转入胞质中,两个ALA 分 子 在 ALA脱 水 酶 作 用 下 合 成 卟 胆 原(PBG),四分子的PBG 在PBG脱氨酶作用下聚合生成线性结构的羟甲基胆色素(HMB),生理状态下肝细胞内的酶催化HMB环化生成尿卟啉原Ⅲ,少部分生成尿卟啉原Ⅰ。尿卟啉原Ⅲ经脱羧酶作用生成粪卟啉原Ⅲ,后经粪卟啉原Ⅲ氧化酶作用转化为原卟啉原Ⅲ,其在原卟啉原Ⅲ氧化酶作用下生成原卟啉原Ⅸ(PP),最后血红素合成酶(即铁螯合酶)催化亚铁离子与PP合成血红素。急性间歇性血卟啉病是最常见的一种类型,AIP 是由于血红素生物合成途径中PBG脱氨酶缺乏以至于血红素前体物质ALA 及PBG的合成增多,蓄积在肝脏中,随血液循环进入组织,尿中排泄增多。血红素合成途径中的第一个关键酶 5-氨基酮戊酸合成酶(ALAS-1)的诱导或血红素合成需求量增大而肝脏 ALAS-1反馈抑制减弱可导致 ALA、PBG的蓄积合成明显增多,导致临床症状急性发作[2]。AIP 的年发病率为(2~3)/10万,多见于成年女性,饮酒、感染、紧张、焦虑、月经来潮、苯巴比妥类药物等均可诱发或加重 AIP[3]。不同诱发因素都是通过直接或间接激活血红素合成过程中肝脏细胞内ALA 合成酶活性,引起卟啉及其前体物质在组织、循环中蓄积。女性患者发病机制尚不清楚,黄体酮、细胞色素酶P450、部分神经递质都可引起肝脏异常代谢,诱发卟啉病急性发作,导致神经功能紊乱及肝脏损伤[4]。本文两例均无诱因,例1性激素检查睾酮升高,查阅相关文献,尚无血卟啉病性激素异常的相关报道。
AIP临床表现各异,以腹痛和神经精神症状为主[5],后者可累及中枢、外周、自主神经系统。(1)腹痛:是AIP患者最常见的一种症状,其特点是疼痛较重而弥散,呈进行性加重的腹部绞痛,可伴腹肌紧张、压痛、反跳痛,伴或不伴膀胱区及后背部放射痛,中腹部较常见,持续时间数小时至数天不等。常伴恶心、呕吐、腹肌紧张、便秘等,便秘可能与膀胱肌麻痹尿潴留有关。(2)中枢神经系统症状:意识障碍、癫痫、焦虑、抑郁、失眠、幻觉等。癫痫发病率高达5%,可能由低钠血症或不恰当用药引起。(3)周围神经系统症状:以运动症状为主,多首先累及双上肢近端,表现为肌无力肌痛,后可渐累及四肢、延髓肌,致四肢瘫痪、呼吸无力,需呼吸机辅助通气,严重者可致死亡。感觉症状较少见,表现为神经痛、远端感觉缺失、近端肢体“穿泳衣”感。在急性发病期,腹痛常伴有一过性的上述肢体感觉症状。此外,脑神经病变多发生在并发神经系统症状的血卟啉患者,发生率达75%,且通常发生在肢体及躯干受累后,常累及面神经、迷走神经,三叉、舌下、副神经及动眼神经也可受累。视神经萎缩、眼肌麻痹、面神经瘫痪、吞咽困难、声带麻痹等也有报道。(4)自主神经症状[6]:包括心动过速、心律不齐、高血压、尿潴留、低热、出汗、脱水、电解质紊乱等。电解质紊乱多表现为低钠血症,发病率高达30%,推测原因,可能是由于下丘脑功能受损、抗利尿激素分泌胃肠道丢失过多引起[7]。经典的腹痛、神经病变、精神异常三联征可发生于50%的急性发作患者中。单纯出现神经精神症状而无腹痛症状者较少见,但的确存在单独出现神经损害、脑病及精神症状的报道[8]。本文例1临床表现较典型,腹痛、癫痫、易激惹典型AIP三联征表现,四肢肌力减低、腱反射消失、痛觉过敏、呼吸无力等周围神经症状,低血钠、高血压、窦性心动过速等自主神经紊乱。例2以腹痛为主要表现,并窦性心动过速,无精神神经系统表现。神经系统症状女性多见于男性,并且在青春期前及绝经后少见[9]。目前关于卟啉致神经病变主要有两种假说:(1)卟啉类及其前体物质过度蓄积直接损害神经组织;(2)由于血红素生物合成途径中相关酶缺乏导致血红素生成减少,细胞能量缺乏致轴突退化[10]。
血卟啉病的神经系统影像学表现及机制:主要有三种表现形式:(1)皮质与皮质下白质病变:两侧额叶、顶叶、枕叶皮质及皮质下白质(白质为主)斑片样长 T1、长 T2信号影,DWI像上多呈低信号[11],ADC图呈高信号,FLAIR像呈较高信号,病灶可融合。病变多发、多成对称性改变,部分病变存在可逆性,与可逆性大脑后部白质脑病综合征的病变分布和特点相似,二者可能存在相同的病理生理学机制[12]。也可能由于血卟啉病患者机体中血红素缺乏导致NO产生减少(NO为一种重要的血管舒张因子)导致血压升高和脑血管收缩[11]。(2)脑深部核团病变:双侧尾状核头及豆状核、丘脑等深部灰质核团对称性病变,病变呈长T1长 T2信号,FLAIR 像为高信号,DWI信号稍高,ADC像呈高或低信号[11]。可能与卟啉及其代谢前体物质对神经的毒性作用及频繁痫性发作导致的缺氧性脑病有关。(3)脑缺血性病变:急性发作患者中,出现可逆与不可逆性缺血改变。可能与血管痉挛、血管收缩等有关。本文例1表现为皮质与皮质下白质病变,前后复查头部磁共振,病变呈现可逆性改变。例2表现为点灶状缺血性改变。
诊断及鉴别诊断:血卟啉病的诊断需综合上述临床、影像表现及相关辅助检查[10]:AIP最重要的快速诊断方法是随机尿PBG含量的定性及半定量测定。尿液检测:ALA、PBG、尿卟啉明显升高;血清、粪便:卟啉类物质轻度增高或正常。相关DNA基因突变位点检测是诊断的金标准。AIP的遗传方式是常染色体显性遗传,目前报道的有关AIP 基因突变位点有300多个,HMBS基因c. 518G>A(p. Arg173Gln)为已知致病性变异,但基因外显率低,部分患者携带基因但不发病,基因型和表型无必然关联,即某些突变并不代表疾病严重程度,疾病严重程度还受环境和其他因素影响[13]。即使携带同一致病基因的患者,其临床表现也可有很大差别[14]。尿液实验:尿液经加热、加酸或暴晒后变红甚至发黑。AIP患者肝脏 PBG 脱氨酶缺乏,多余的PBG经肾脏排出,PBG在尿液中可转化成尿卟啉,可能导致尿液中尿卟啉、粪卟啉含量升高,最终卟啉类化合物及卟吩胆色素形成使尿液颜色加深。本文2例AIP患者及其父亲验证了上述观点。双胞胎患者及其父亲均为同一基因型,符合AIP常染色体显性遗传,但三者临床表现完全不同,例1病情重并进展迅速,例2病情较轻,高糖治疗后症状迅速缓解,患者父亲自述无明显临床症状,为AIP无症状携带者。追问患者父亲,其兄弟姐妹及父母均无患者类似表现,因目前广大民众对遗传病的讳莫如深,未能对其家系进行基因检测。应做好相关宣传教育,对发现血卟啉病患者的家系进行筛查,随访基因携带者,避免诱因,出现症状及时治疗。最为重要的是对携带者进行产前诊断,优生优育,阻断家系遗传。
鉴别诊断:由于本病发病率低、较罕见且无特异性临床表现,误诊率高,应做好鉴别诊断。(1)患者急性间歇性腹痛发作时,常被误诊为急性阑尾炎、急性胰腺炎、消化性溃疡、胆结石、泌尿系结石等。急腹症发作有各自特有的临床特点,AIP腹痛发作时常伴有尿液实验阳性[15]。(2)单纯出现神经精神症状而无腹痛症状者较为少见,但确存在单独出现神经损害、脑病及精神症状的报道,此时易与吉兰巴雷综合征、颅内病变等相混淆。AIP神经传导研究表明以轴突损害为主,而吉兰巴雷综合征多为脱髓鞘病变[10]。颅内病变:颅内感染等可通过影像学及脑脊液检查相鉴别,AIP患者脑部多见可逆性后部白质病变。
治疗:(1)明确并避免诱因,如相关药物的应用、饮酒、饥饿、感染等。(2)对症治疗:腹痛者可使用阿片类止痛剂;心动过速、高血压可使用倍他洛克;恶心、呕吐可使用奥氮平、劳拉西泮等,但多潘立酮、胃复安等应慎用;尿潴留者可导尿;癫痫发作可静脉使用地西泮、左乙拉西坦,一般癫痫呈一过性发作,不需要维持治疗[16];(3)血红素疗法:缓解症状快、效果好,常见不良反应有轻度凝血功能异常、血栓性静脉炎、过敏性休克等。但目前我国大陆无血红素制剂上市。(4)高浓度葡萄糖疗法:高浓度葡萄糖(300 g~500 g)/d用于卟啉病轻度发作或是无条件使用血红素时,可减少卟啉前体在组织中沉积,并可游离部分沉积于组织中的卟啉前体,但症状缓解不明显者需要尽快使用血红素治疗,同时应积极防治低血钠等电解质紊乱。(5)激素疗法:孕激素、雌激素用于治疗卟啉病疗效不明显,可诱发卟啉病,指南并不推荐使用。(6)肝移植疗法:治疗效果差、生活质量差、反复发作的患者,可考虑肝脏移植治疗,严重的AIP患者肝脏移植成功后临床症状明显改善[17],但有相关移植风险。从本文2例的治疗效果可见高糖治疗对症状较轻者疗效显著,但对于重症者疗效欠佳。伴有神经症候群的患者预后不良,应早发现,早治疗,高糖治疗效果不显著时及时应用血红素及其他疗法。
AIP治疗前景:重组人卟胆原脱氨酶(recombinant human-HMBS-enzyme,rh-HMBS) 是未来治疗卟啉病的方法之一,rh-HMBS 以增加患者尿中卟啉物质排泄降低其在血中浓度而达到治疗目的。如何使rh-HMBS 进入肝细胞并恢复肝细胞内 HMBS 的正常活性及血红素的正常合成是当前阶段研究重点[18]。RNA干扰治疗在动物研究中证实有效,但临床试验效果不佳[19]。腺病毒介导的基因治疗I期临床试验效果不显著[20]。
通过报道上述两例同基因型同卵双胞胎姐妹同患血卟啉病的不同临床表现,以期提高对血卟啉病认识。急性间歇性血卟啉病为常染色体显性遗传,相同基因型可有不同表型,预后亦有很大差别,应早发现,早治疗,避免诱发因素。高糖治疗对症状较轻者疗效显著,症状较重者需及时应用血红素疗法。
