误诊为IHH的单纯男性化型男性21羟化酶缺陷症一例
2019-04-03刘艳霞栗夏莲
刘艳霞,高 月,栗夏莲
郑州大学第一附属医院内分泌科;河南省内分泌代谢中心 郑州 450052
21羟化酶缺陷症是由于21羟化酶缺陷导致肾上腺皮质激素合成障碍的最常见的先天性肾上腺皮质增生症。成年男性21羟化酶缺陷症患者症状往往不典型,多因不育或肾上腺肿物就诊。肾上腺来源的过量雄激素及中间代谢产物的蓄积可长期抑制下丘脑-垂体-睾丸,致使黄体生成素(LH)和促卵泡刺激素(FSH)水平下降,睾丸不能发育成熟,易被误诊为低促性腺激素性性腺功能减退症(isolatedhypogonadotropichypogonadism,IHH),导致误诊误治。现报道一例误诊为IHH的21羟化酶缺陷症,以期提高对这两种疾病的认识和诊断水平。
1 病例资料
1.1一般资料患者,男,30岁,因婚后未避孕不育8 a,至外院查性激素六项: LH 0.007 mIU/L(正常参考值1.000~12.500 mIU/L,后文括号内均为正常参考值), FSH 0.012 mIU/L (1.000~12.000 mIU/L), 雌二醇(E2) 135 ng/L(<75 ng/L), 泌乳素(PRL) 137.9 mIU/L (42.5~414.0 mIU/L), 睾酮(T)3.5 μg/L(2.8~12.0 μg/L),孕酮(P)14.7 μg/L(0.1~2.0 μg/L)。睾丸超声示:左侧睾丸22 mm×14 mm×14 mm,右侧24 mm×14 mm×17 mm,轮廓清晰,包膜光滑。垂体MRI示:垂体前叶下部片状稍长T2信号,增强扫描未见明显异常。精液分析:排精量4.6 mL(>2 mL),酸碱度pH7.4 (pH7.2~8.0),精子活动率0%(>60%), 精子活力不正常,精子密度 0×106个/mL(>20×106个/mL),精液离心镜检未找到精子。睾丸穿刺活检:穿刺液0.7 mL,高倍镜下未见精子。达比佳激发试验(静脉注射戈那瑞林60 min)LH 1.359 IU/L。肝功能、肾功能、电解质均正常。诊断为特发性IHH,拟行促性腺激素替代治疗。
经我院会诊,认为患者LH与FSH水平低,T水平正常,未服用任何药物,与IHH临床特点及体征不相符,考虑睾酮为肾上腺来源。进一步完善病史:患者性生活可正常完成,每周2~3次;体格检查示嗅觉正常,有胡须,身高160 cm,体重64 kg,体重指数25.0 kg/m2,血压120/85 mmHg(1 mmHg=0.133 kPa),阴毛浓密呈菱形分布、色深、未到脐,阴茎长度约9 cm,双侧睾丸约4 mL(Prader睾丸计)。追踪生长发育史:足月顺产,头先露,出身后无拒食、呕吐,6~8岁时身高较同龄人高,9岁时阴茎有不自主勃起现象,未就诊。父母已故,爱人体健,父母及两个妹妹均体健,两妹自然受孕。既往史、个人史无特殊。肾上腺CT(图1)示双侧肾上腺增粗,见等密度结节影,边缘不整,密度均匀。
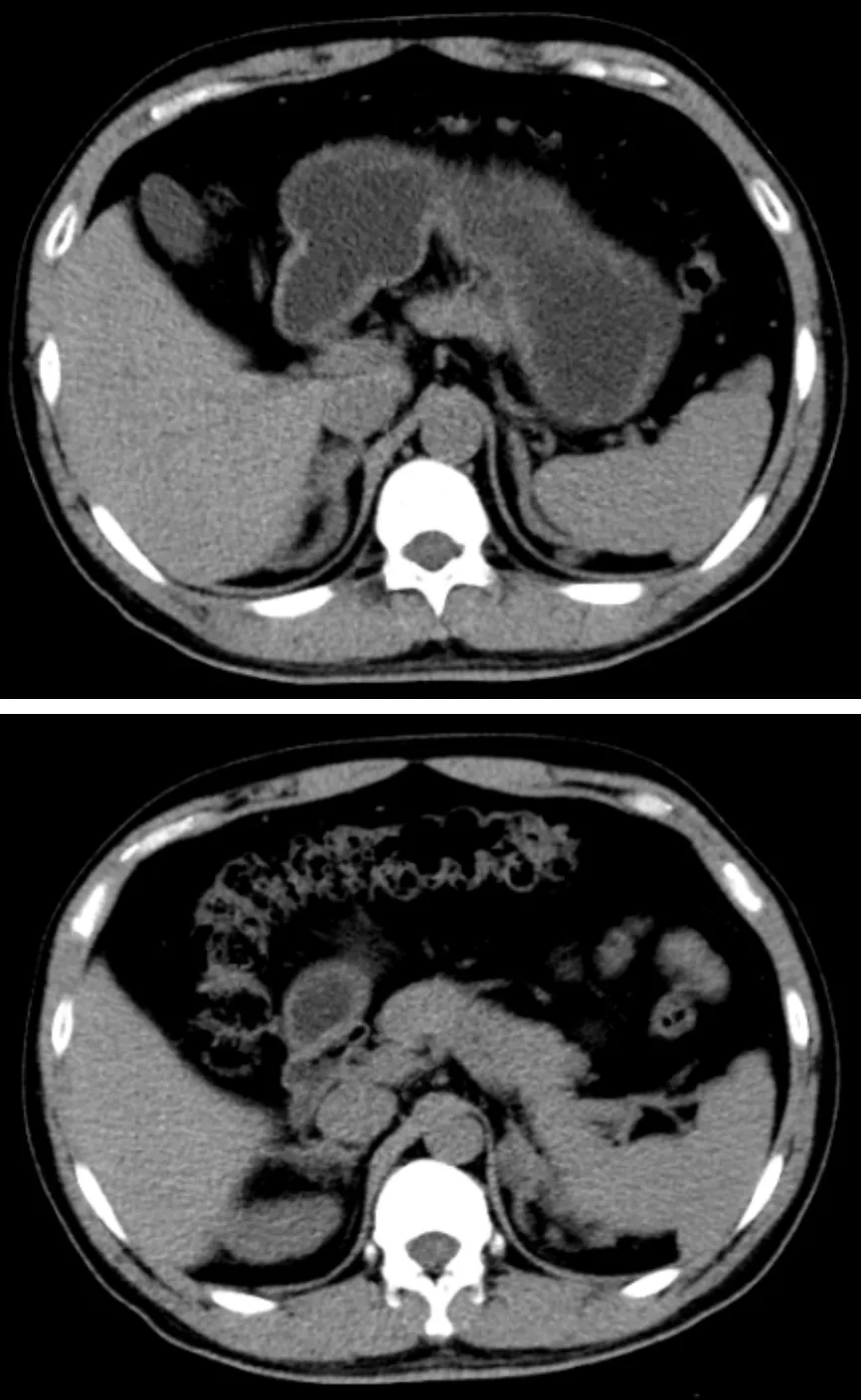
图1 患者肾上腺CT平扫表现
1.2实验室检查我院采用全自动生化仪测定肝肾功能、血电解质等,化学发光法测定LH、FSH、T、硫酸脱氢表雄酮(DHEAS)、雄烯二酮(AD)、肾上腺皮质激素(ACTH)、皮质醇(COR)等内分泌激素。性激素六项:FSH为0.16 mIU/L(0.95~11.95 mIU/L),LH为0.01 mIU/L(1.14~8.75 mIU/L), P为5.74 μg/L(<0.10~0.20 μg/L),E2为31 ng/L(11~44 ng/L),T为4.98 μg/L(1.42~9.23 μg/L),PRL为14.18 μg/L(5.18~26.53 μg/L);其他内分泌相关激素: DHEAS为376 μg/dL(80~560 μg/dL,1 μg/dL=0.01 mg/L),AD>10.0 μg/L(0.6~3.1 μg/L),17-羟孕酮(17-OHP)为47.88 μg/L(0.61~3.34 μg/L);肾素活性卧位为1.18 μg/(L·h)[(0.15~2.33)μg/(L·h)],立位为5.54 μg/(L·h)[0.1~6.56 μg/(L·h)];醛固酮卧位为243.1 ng/L(30.0~160.0 ng/L),立位为243.1 ng/L(30.0~160.0 ng/L)。ACTH-COR节律见表1。

表1 ACTH-COR节律
1.3基因检测获得患者知情同意后,抽取患者静脉血5 mL,EDTA抗凝,应用过柱法检测试剂盒(美国AIAGEN公司)抽取外周血DNA,送至上海韦翰斯生物医药科技有限公司用ABI3700荧光自动测序仪测序,用Primer 5.0设计特异性引物,结果与NCBI网站的CYP21基因序列(Gene ID为1589)进行比对。Sanger测序结果(图2)显示,患者CYP21A2基因第6外显子存在杂合错义突变c.518T>A(p.I173N)。
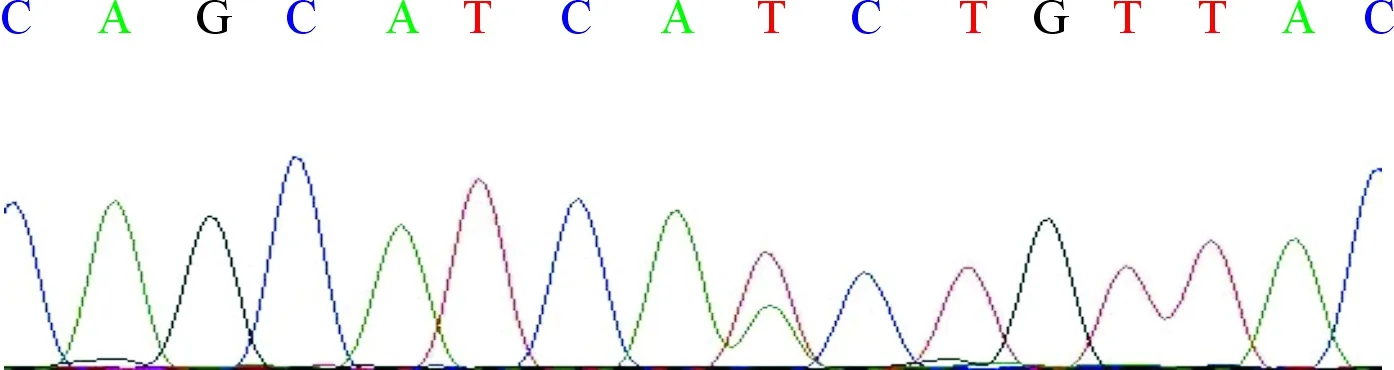
图2 患者Sanger测序峰图
2 讨论
该例患者为30岁男性,婚后不育8 a,性生活正常(未使用药物),内分泌检查提示LH、FSH低,睾酮正常,达必佳试验LH可以上升到1.359 IU/L,故外院诊断为IHH。但患者身材偏矮,阴毛浓密呈菱形分布,阴茎长度正常,实验室检查睾酮正常,考虑肾上腺为睾酮的另一个来源,故进一步评估肾上腺相关激素,结果显示DHEAS、AD、17-OHP和ACTH均升高,基因检测发现CYP21A2基因第6外显子存在杂合错义突变:c.518T>A(p.I173N),故最终诊断为单纯男性化型21羟化酶缺陷症。
21羟化酶缺陷症是由于在肾上腺皮质激素合成途径中21羟化酶缺陷导致的常染色体隐性遗传病,是先天性肾上腺皮质增生症最常见的类型,根据酶缺乏程度分为失盐型、非经典型和单纯男性化型[1]。失盐型患者21羟化酶完全缺乏,皮质醇和皮质酮合成均出现障碍,胎儿期起病,可出现高钾血症、低钠血症、代谢性酸中毒、低血糖症等代谢紊乱症状,甚至肾上腺危象。非失盐型患者由于症状不典型,就诊率低。成年男性21羟化酶缺陷症患者生育能力下降,常因婚后不育及睾丸肿物而就诊[2],临床极易误诊。
特发性IHH是临床上造成男性性发育异常的疾病之一,典型表现为小阴茎、睾丸体积小(1~3 mL)、第二性征不发育或发育不全,骨骺闭合延迟呈现“宦官样”体征,促性腺激素水平低或正常,且睾酮水平≤3.47 nmol/L[3]。单纯男性化型21羟化酶缺陷症患者21-羟化酶不完全缺乏,醛固酮合成正常,雄激素过量,男性可表现为外周性性早熟,阴毛提早出现,阴茎增大且容易勃起,但睾丸体积仍为青春发育前大小,体格发育过快,骨龄提前,成年后身材偏矮,皮肤黏膜色素沉着,肾上腺来源的睾酮可使第二性征发育,维持性生活。所以详细的病史采集和体格检查是鉴别二者的重要依据之一。
男性21羟化酶缺陷症患者不育原因之一为CYP21活性降低或消失,P和17-OHP不能被转化为11-去氧皮质酮,COR合成减少,对下丘脑和垂体的反馈抑制作用减弱,ACTH分泌增加,刺激肾上腺皮质(主要为束状带)增生,导致过量17-OHP与P产生。过量的17-OHP和P一部分在17,20-碳裂解酶作用下进入雄激素合成途径。过量的雄激素可在外周通过芳香化酶的作用部分转化为雌激素,过量的雄激素、雌激素及17-OHP、P、AD等中间代谢产物共同长期抑制下丘脑-垂体-睾丸,反馈抑制LH和FSH;睾丸不能发育成熟,表现为睾丸细小、坚硬,前列腺发育不良,曲细精管和睾丸间质细胞发育异常、精子生成障碍,从而导致不育[4]。因肾上腺雄激素的作用,男性第二性征存在,阴茎正常或增大,有时甚至表现为毛发增多,但患者身材偏矮,多伴有中心性肥胖、高血压、胰岛素抵抗甚至2型糖尿病等代谢综合征的表现,也影响患者生育能力[5]。睾丸肾上腺残余瘤是导致成年男性21羟化酶缺陷症不育的另一重要因素。增高的ACTH和血管紧张素Ⅱ(AT-Ⅱ)刺激睾丸内肾上腺皮质残余细胞增生,从而发生睾丸肾上腺残余瘤,压迫睾丸网导致曲细精管梗阻,其分泌的肾上腺类固醇类激素对睾丸间质细胞和生殖细胞有生殖毒性,严重者可致不可逆的睾丸功能损害[6]。Bouvattier等[7]统计了2011至2014年间219例男性非经典型21羟缺陷症患者,42%有少精症或无精症,合并肾上腺残余瘤的患者少精和无精发生率达70%。
21羟化酶缺陷症部分患者需终生口服肾上腺皮质激素替代治疗[2],缓解COR和醛固酮分泌不足的症状,抑制肾上腺来源的过量雄激素;糖皮质激素强化治疗可提高生育功能受损患者的生育能力[8]。国内外个案报道[9-10]显示合并低ACTH患者的生育能力是可逆的,经短期治疗后均可恢复生精能力。肾上腺皮质激素治疗失败可考虑联合应用促性腺激素治疗促进精子生成及辅助生殖技术。
综上所述,成年男性21羟化酶缺陷症患者症状不典型,应详细询问生长发育史、评估临床体征,以免误诊。
