Relationship between pore structure and hydration activity of CaO from carbide slag☆
2019-02-09JunqiangZhangShuZhangMeiZhongZhiWangGuoyuQianJunhaoLiuXuzhongGong
Junqiang Zhang,Shu Zhang*,Mei Zhong,Zhi Wang,Guoyu Qian,Junhao Liu,Xuzhong Gong,4,*
1 Key Laboratory of Green Process and Engineering,National Engineering Laboratory for Hydrometallurgical Cleaner Production Technology,Institute of Process Engineering,Chinese Academy of Sciences,Beijing 100190,China
2 College of Materials Science and Engineering,Nanjing Forestry University,Nanjing 210037,China
3 Key Laboratory of Coal Clean Conversion&Chemical Engineering Process(Xinjiang Uyghur Autonomous Region),College of Chemistry and Chemical Engineering,Xinjiang University,Urumqi 830046,China
4 University of Chinese Academy of Sciences,Beijing 100190,China
Keywords:Carbide slag CaO Hydration activity Sintering Pore volume fraction
ABSTRACT CaO needs to show high activity to be used as Ca-sorbent and slagging agent.Hydration activity is an important characteristic to evaluate the activity of CaO.In this study,carbide slag from polyvinyl chloride(PVC)industry was utilized as precursor for preparing high activity CaO.The roles of crystallite grain,average pore diameter(APD)and volume fraction of pore <200 nm in diameter(VF200)in hydration activity of CaO from carbide slag(CS-CaO)were respectively investigated.The hydrolysis kinetics model of CaO shows a three-dimensional spherically symmetric diffusion model(D4),which suggests that hydration activity was mainly associated with APD and VF200 of CS-CaO with limited correlation to the crystal size.Specifically,the hydration activity of CS-CaO is increased with increasing VF200,while decreased with increasing APD.Under the invariable calcination temperature,the core-shell structure formed by the addition of graphite or CaCO3to CS effectively inhibits the sintering of CS-CaO and improves VF200.Consequently,the hydration activity of CS-CaO increased from 22.79°C·min−1 to 27.19°C·min−1 and to 29.27°C·min−1,with addition of 5%graphite or 5%CaCO3into carbide slag,respectively.
1.Introduction
Carbide slag,composed of 85%Ca(OH)2,is a by-product of the process of acetylene (C2H2)production from calcium carbide (CaC2)in PVC industry[1].It is estimated that around 5.6×107tons of carbide slag are produced annually in China,however,only 40% are being used efficiently [2].Disposal of massive amounts of carbide slag as waste is neither environmentally nor economically friendly.On the one hand,there is a high demand on CaO usage as a trapping agent[3-5]and platform materials[6-8]etc.However,CaO production is in shortage due to the restriction of limestone exploitation[9-13].Therefore,CaO preparation from carbide slag is considered as an effective approach for the sustainable development of PVC industry.Unfortunately,the performance of CaO from carbide slag still needs to be improved,especially for its hydration activity,because the application of CaO depended on its hydration activity[14].
Powdery CaO is widely used as a Ca-based absorbent for acidic waste gases such as CO2[15-19],SO2[20,21],and HCl[22-26],as well as a slag-refining agent for the steel industry [27-29].However,those applications require CaO to be reactive,which could be characterized by hydration activity [30,31].Consequently,enhancing hydration activity is the key for increasing the value of the powdery CaO.
In nature,the hydration activity is the reactivity between CaO and water,which was similar to the reaction of CaO with CO2(carbonation reactivity or CO2capture capacity)[32,33].Previous research indicated that the particle size,the content of free CaO and pore structure all play important roles in hydration activity [16,32].For example,CaO with the middle-sized particles(150-250 μm)shows the highest activity[34]and carbonation reactivity of CaO was in direct proportion to specific surface area but was inversely proportional to grain size[35-37].In addition,the presence of impurities such as SiO2,Al2O3and MgO,in the limestone could accelerate the sintering of CaO particles,leading to the decrease in free CaO content[34,38]and the hydration activity of CaO[4,14].More importantly,it is presumed that the hydration activity of CaO may be related to certain crystal face of CaO crystalline.Some believe that(001)surface of CaO was very reactiveto water[39],while CaO with a highly(111)-oriented plane was also reported to have a high surface energy and thus high hydration activity[40,41].Therefore,the relationship between structure and hydration activity for CaO was still debatable.

Table 1 Chemical components of four different precursors of CaO(wt%)
To improve the CO2capture capacity,several methods have been developed.First,various organic calcium salts were prepared by using organic acids[42-46].After the calcination of the organic calcium salts,the porous CaO sorbents are formed,and thus improving the carbonation reactivity of CaO[47].The production process for solution modification of Ca-based sorbents was simple and convenient for use in practice.Nevertheless,the organic solution modification is still very uneconomical[39].Second,CaO surface doped with alkali metal species could increase basic sites and improve the absorption capacity of CO2[48].Additionally,the dispersing precursors(oxides)used as support materials[49-57]could also hinder the sintering of CaO particles.Third,sepiolite and mixture of biodiesel by-product were added into carbide slag [58,59]to increase the surface area and pore volume of CaO to improve the CO2absorption activity.Although the doping of metal oxide,sepiolite and bio-products improved the absorption capacity of Ca-based sorbents,the formation of complex compounds such as Ca12Al14O33,Ca3Al2O6,and Ca2SiO4as new impurities inevitably hinders the further improvement of its hydration activity.To alleviate the sintering of CaO particles,different calcination styles were carried out to improve the CaO activity,such as high-concentration steam activation [12,60].However,under constant calcination temperature,how to prevent CS-CaO particle sintering was still very challenging.
In this work,calcium hydroxide (CH),calcium carbonate (CC),carbide slag (CS)and limestone (LS)were used as precursors for CaO.The microstructure of CaO was analyzed,and the relationship between pore structure and hydration activity of CaO was revealed.Further,carbon powder(graphite)and CaCO3powder were used to directional regulation the sintering structure of CaO particles under constant calcination temperature to increase hydration the activity of CS-CaO.

Table 2 Preparation conditions from four various precursors of CaO
2.Experimental
2.1.Preparation of samples
Limestone (LS)was taken from CaC2production enterprises in Shandong Province,China.The carbide slag(CS)was sampled from a chlor-alkali plant located in Shandong Province,China.The chemical components of the LS and the CS were analyzed by X-ray fluorescence(XRF)as shown in Table 1.CaCO3and Ca(OH)2were named by CC and CH,respectively.(CC ≥99.0%,AR,XiLong Scientific,China;CH ≥99.0%,AR,Aladdin Industrial Corporation,China).Graphite was highly pure grade(99.9%)with particle size <0.125 mm.
CS is subjected to flotation(or hydro cyclone)prior to calcination to remove manifest impurities[61].The industrial CS was poured into a Teflon beaker(5 L)and sufficiently stirred.The well-mixed slurry was filtered through a 100 mesh(0.15 mm)standard sieve and the filtrate was collected with vacuum filtration.
The LS is crushed to 5-20 mm by the jaw crusher(PE60X100,Tencan powder,China)before calcination,and the error of the coarse sample content is eliminated.All calcined samples should be ground and sieved(100 mesh:0.15mm)to prevent the presence of large particles of material during the experiment.
The current industrial production of CaO is based on the use of massive limestone calcined in a shaft furnace(moving bed)[62].Therefore,this experiment used a cylindrical corundum crucible (50 mm in diameter and 100 mm in heights)to simulate CaO production,as shown in Fig.1.All samples were heated in an air atmosphere at a furnace heating rate of 10°C·min−1to a specific target temperature for 60 min (ensuring the complete conversion of the precursor into CaO),subsequently cooled to room temperature for sampling.Sample preparation conditions are shown in Table 2.The CaO samples prepared from CH,CS,CC,and LS were named by CH-CaO,CS-CaO,CC-CaO,and LS-CaO,respectively.

Fig.1.Picture of samples before(a)and after calcination(b).1.Agitator,2.Electrode,3.Agitator rotator,4.Controller,5.Vessel.
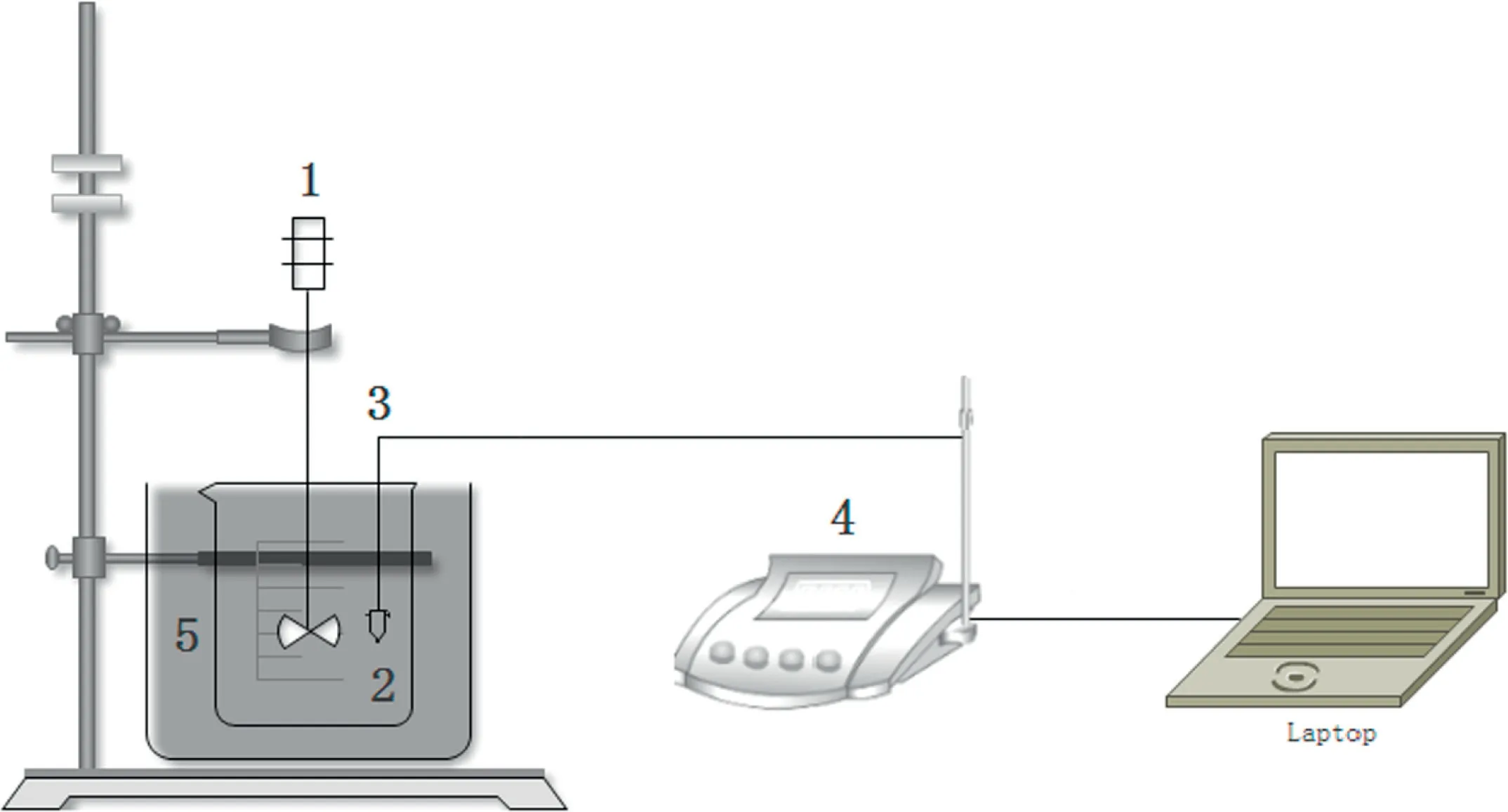
Fig.2.Hydration activity testing device for powdery CaO.
2.2.Hydration activity(α)test
The hydration activity tests of the samples were carried out by following Canadian national standard method[63],featuring a thermocouple and computer operated under room temperature,as shown in Fig.2.The thermocouple online coupling Labx software was applied in this device to acquire reaction temperature instantaneously.
Test steps were as follows:(1)225 ml of deionized water was placed in a 1000 ml container with thermal insulation properties.(2)The sample used was ground to a certain size and sieved with 100 mesh sieves(0.15 mm),and the particle size should be as uniform as possible.(3)Reaction is processed at the speed of 300 r·min−1(4)Test the temperature of the solution until the temperature of it reach to maximum value.(5)75 g (weighed in advance)samples were added to the insulation container.The temperature was recorded every 1 s until the solution temperature began to drop,from which the maximum temperature(Tmax)was obtained.(6)Calculation of CaO hydration activity is as shown in Eq.(1).

where α is the hydration activity of CaO(°C·min−1);T0and Tmaxare the initial temperature(°C)and maximum temperature(°C).Each sample was tested five times to obtain average value as the final hydration activity of CaO.
2.3.Characterization of samples
The sample composition was detected by an AXIOS-MAX X-ray diffraction f(XRF).X'Pert PRO MPD X-ray diffraction(XRD)was used to detect the phase compositions and crystal structures of the specimens.The microstructures of the samples of the sintered parts by a JSM-7001F field emission scanning electron microscopy(SEM).The element distribution in the surface of the specimen was detected by INCA X-MAX X energy dispersive X-ray(EDX).The pore structure parameters of the samples were determined by Autopore IV9500 automatic mercury porosimetry.A thermo-gravimetric analyzer(Pyris 1 TGA,PerkinEImer)was used to investigate mechanism of carbide slag conversion to CaO.The test was conducted at a temperature of 20 °C·min−1from room temperature to 900°C under an air atmosphere of 40 ml·min−1.The heating program of high temperature steam thermogravimetry:after the temperature is raised to 250°C at 20°C·min−1,the water vapor is introduced,and the temperature reaches 900 °C to stop collecting data.The atmosphere was H2O and Ar in turn,both of which had a flow rate of 200 ml·min−1.
3.Results and Discussion
3.1.Hydration activity of CaO from different precursors
Fig.3 shows the hydration temperature rising curves of CH-CaO,CSCaO,CC-CaO and LS-CaO at different temperatures.The slope of these curves first increases and then decreases with increasing calcination temperature.It is obvious that the highest temperature from hydration reaction of CH-CaO and CC-CaO is 85°C at 100 s,compared to 75°C from hydration reaction of LS-CaO and CS-CaO at a longer reaction time(>140 s).
According to the temperature rising curves of CaO hydration reaction,the hydration activity of CaO from different precursors can be calculated by using Eq.1,as shown in Fig.4.The hydration activity increases and then decreases with increasing calcination temperature.The optimal calcination temperature of CH and CS was 900°C,while the optimal calcination temperature of CC and LS was 1100 °C.The highest hydration activity of CH-CaO(αCH-CaO)and CS-CaO(αCS-CaO)at 900°C was 32.72°C·min−1and 22.79°C·min−1,respectively.The highest hydration activity of CC-CaO (αCC-CaO)and LS-CaO (αLS-CaO)at 1100 °C was 37.9 °C·min−1and 18.42 °C·min−1,respectively.It should be noted that αCS-CaO(22.79°C·min−1)is 23.73%higher than αLS-CaO(18.42°C·min−1).

Fig.4.Effect of calcination temperature on CaO hydration activity prepared from different precursors(60 min).
The difference in hydration activity of CaO from different precursors can be attributed to different contents of CaO and impurities in the precursor.As shown in Table 1,on the one hand,impurity resulted in the decrease in CaO content.On the other hand,impurity improved the sintering of CaO particles.Therefore,the high content in CaO would lead to the high hydration activity.The hydration activity of CS or CH increases with increasing calcination temperature due to the decreasing contents of Ca(OH)2.However,the sintering of CaO particles also becomes increasingly significant after the calcination temperature reached 900°C,and thus higher calcination temperature lead to lower activity.The inherent impurities (SiO2,Al2O3,Fe2O3,MgO,etc.)in the CS will react with the free CaO to produce the inactive CaSiO3and(CaO)12(Al2O3)7[9].Therefore,the hydration activity of CH-CaO was higher than that of CS-CaO while LS-CaO has the lowest hydration activity.Comparison with LS-CaO,αCS-CaOis higher than αLS-CaOdue to the different contents of the impurity(Table 1)and thus different decomposition temperatures(500°C and 900°C)[56,57].
3.2.Relationship between structure and hydration activity of CaO

Fig.5.XRD pattern of CaO prepared from(a)CH,(b)CC,(c)LS and(d)CS(60 min).
XRD analysis of the samples after calcination at 600-1200 °C is shown in Fig.5.As shown in Fig.5(a),there is a small amount of Ca(OH)2(2θ ≈18°)for CH at low temperature.With the increase of temperature,Ca(OH)2is converted into CaO,and the diffraction peak intensity for CaO increased gradually.The 32.26°,37.41°,and 53.92°of diffraction angles respectively corresponded to the(111),(200)and(220)diffraction peaks of CaO.The diffraction patterns in Fig.5(b)and(c)show that a portion of the CC or LS (2θ=29.42,39.45,47.51,48.51°)is still present at 1000 °C until it is completely decomposed into CaO at 1100°C.
Fig.5(d)shows that a small amount of Ca(OH)2and CaCO3were found at range of 600 to 800°C.It can be inferred that CS itself contained CaCO3.The change in XRD pattern of CS after calcination at 800°C is exactly the same as that of CH.Therefore,the optimal calcination temperature for CH and CS is 900 °C,while the optimal calcination temperature for CC and LS is 1100°C.
The characteristic peak intensity can reflect grain size of CaO.The crystal size was calculated by Scherrer formula as shown in Eq.(2).

where,κ is Scherrer constant(κ=0.89 for the CaO cubic structure),λ is the X-ray diffraction wavelength of 0.154056nm.The β is full width at half-maximum(FWHM).Thus,it iswhereis the FWHM for each peak,and d=2 because of the peak shape conform approximately to a Gaussian distribution[39].
Relationship between hydration activity and crystal grain of CaO prepared from different precursors at different temperatures is shown in Fig.6.Obviously,the crystal grain is mainly concentrated at the range of 64 to 100 nm.The larger the CaO crystallites are,the smaller the crystal lattice distortion is.The low surface free energy of the crystal was beneficial to structure stability,resulting in low hydration activity for CaO[35].

Fig.6.Relationship between hydration activity and crystal grain of CaO prepared from different precursors.

Fig.7.Morphology of CaO from(a)CH at 900°C,(b)CC at 1100°C,(c)CS at 900°C and(d)LS at 1100°C(60 min).
As shown in Fig.6,CaO with the smallest crystallite(32-64 nm)did not show the highest hydration activity,while the CaO with middle size crystallite(64-100 nm)showed the highest hydration activity.Compared with CC-CaO,the crystal grain of LS-CaO was larger at the same temperature.For LS-CaO,abundant impurities and high decomposition temperature resulted in significant CaO particle sintering.CC-CaO with middle size crystallite has higher hydration activity than LS-CaO.Likewise,the hydration activity of CH-CaO with middle size crystallite was higher than that of CS-CaO.Therefore,there is no direct functional relationship between the crystallite grain of CaO and its hydration activity.Instead,different precursors CaO jointly have an active grain interval of medium grains(64-100nm).When CaO grains are in this interval,its hydration activity is strongest.
As shown in Fig.7,the CH-CaO and CC-CaO particles are mainly arranged in a lamellar structure.Compared with CH-CaO and CC-CaO,the sintering behavior of CS-CaO and LS-CaO is more significant because impurities form low melting point compounds at high temperatures,such as CaSiO3,Ca2SiO4,(CaO)12(Al2O3)9[4].Consequently,the pore structures were destroyed,which could also be certified by a large number of small pore structures for CH-CaO,CC-CaO and CS-CaO.The sintering agglomeration of LS-CaO is the most remarkable withe few pores left in the LS-CaO.
3.3.Hydrolysis kinetics model of CS-CaO
The hydration activity of different precursors CaO may be affected by the hydrolysis kinetics of CaO.Therefore,it is necessary to investigate the hydrolysis kinetics of different precursors of CaO.The reactionkinetic model has a one-dimensional diffusion model,a twodimensional diffusion model,and a three-dimensional diffusion model[64,65].The detailed classification is shown in Table 3.In this work,the hydration conversion rate (x)of CaO was obtained from the thermo-gravimetric curve under steam(Fig.8),and then optimized by various kinetic equations to obtain the optimal hydration kinetics model of CaO.

Table 3 Gas-solid reaction kinetic model[64,65]

Fig.8.Thermo-gravimetric analysis of(a)CS,(b)CS with 5%graphite and(c)CS with 10%graphite under steam.

where,m0and m were the weights of samples at initial and reaction,respectively.
Assume that the hydrolysis reaction model of CaO is Eq.(4).


Eq.(3),Eq.(4)and Eq.(5)were simplified to obtain Eq.(6):

The reaction rate at this stage is mainly affected by the volume fraction of pore <200 nm in diameter (VF200)in the reactant [23],and therefore,the pore structure of CaO particle is investigated by mercury intrusion analysis.The results showed that the average pore diameter(APD)of CH-CaO,CC-CaO,CS-CaO,and LS-CaO was 594.7nm,766.1nm,111.6nm,and 1375.4nm,respectively(Fig.10).The VF200 is calculated after integrating the pore size distribution,as shown in Fig.11.The order of VF200 is VF200LS-CaO<VF200CS-CaO<VF200CH-CaO<VF200CC-CaO.Clearly,hydration activity was proportional to VF200 and was inversely related to the APD.As described above,the hydration activity of LS-CaO-1100 was only 18.42°C·min−1,because its VF200 was the lowest.In conclusion,how to suppress sintering and regulate the pore structure of CaO has become the focus of this study.
3.4.Improvement of pore structure by graphite/CaCO3and its mechanism
To improve the hydration activity of CS-CaO under the invariable calcination temperature and purification of CS,it is economical and practical to suppress the sintering of CaO particle using local solid waste from CaC2production industry,such as char powder and limestone powder.Calcination of carbide slag with char would generate CaCO3due to CO2formation from char combustion[67].The decomposition temperature of limestone(900°C)is higher than that of Ca(OH)2.Therefore,CaCO3could alleviate the sintering of CaO particles.In this work,pulverized graphite(high purity)and CaCO3(chemistry agent)were used to represent the char and limestone powders respectively to improve the hydration activity of CS-CaO.

Fig.9.Relationship between lnD4 and 1/T.
As shown in Fig.12,the hydration activity of CS-CaO clearly changed with graphite and CaCO3additions at different extents.With the increase of graphite or CaCO3,the hydration activity of CS-CaO increases firstly,and then decreases.It was noted that the hydration activities of CS-CaO added with 5% graphite and 5% CaCO3were 19.32% and 28.44%higher than that of original CS-CaO,respectively.This clearly suggested that graphite and CaCO3as dispersants have inhibited the sintering reactions of CaO particles.
However,when the additive content was more than 5%,the hydration activity of CS-CaO decreased.It is speculated that unreacted graphite or CaCO3would partially remain in the final product of CS-CaO,resulting in a decrease in hydration activity.As shown in Fig.13,graphite and CaCO3could be observed in CS with 10%graphite or 10%CaCO3after the calcination.The CaCO3that failed to decompose into CaO has resulted in a decrease in hydration activity of CS-CaO.

Table 4 Activation energy of different of CaO①
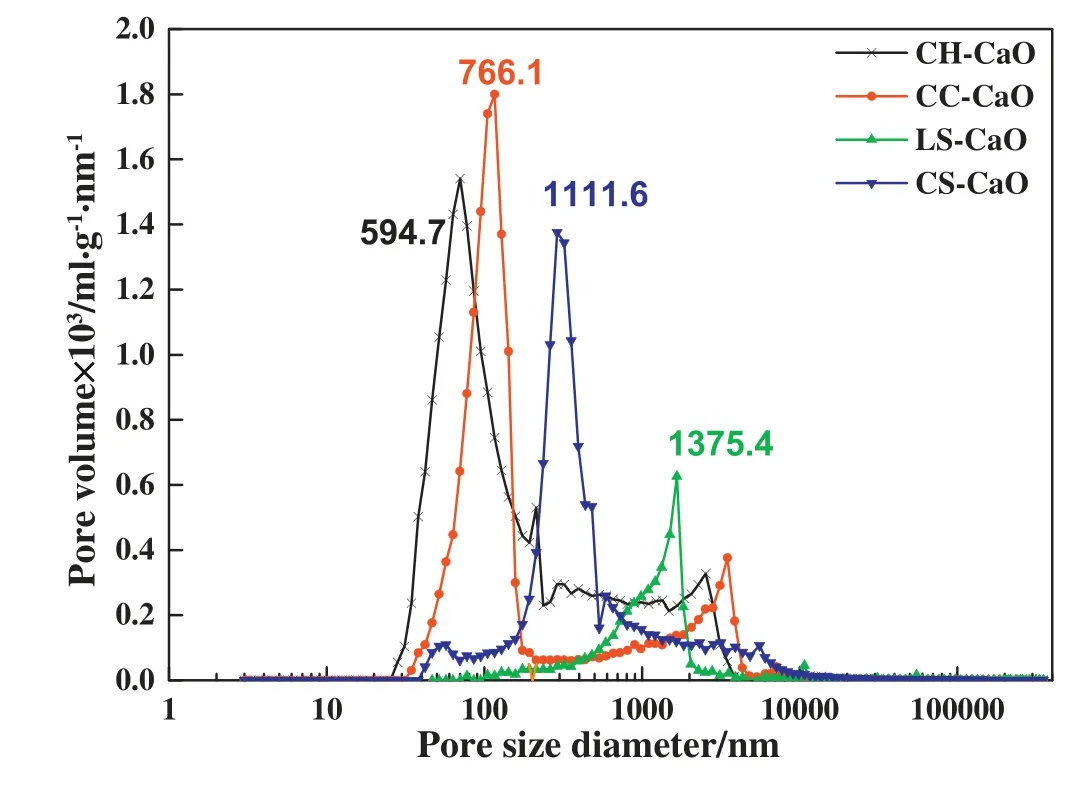
Fig.10.Pore diameter distributions of CaO from different precursors (optimal temperature,60 min).
Fig.14 TG analysis shows that the decomposition of Ca(OH)2in CS was divided into two stages:in the first stage(450°C),most of the Ca(OH)2was converted to CaO,while CaCO3was in-situ generated by the reaction between CO2in air and CaO,and the second stage was the decomposition of generated CaCO3.TG results indicated that the addition of graphite resulted in the increase of CaCO3in CS-CaO.For CS with graphite,core-shell structure CaCO3was in-situ generated by CaO with CO2that generated from graphite combustion.Thus,the CaCO3layer inhibited the sintering of CaO particles.CaCO3was acting as a dispersant to prevent the continuous sintering of the CaO particles.And finally,the CaCO3was also decomposed into CaO.Obviously,with the addition of graphite or CaCO3,the abundance of CaCO3increased,and thus the decomposition temperature range of CaCO3became wide.Therefore,the sintering of CS-CaO particles was alleviated and the hydration activity was improved.

Fig.11.VF200 and APD as a function of hydration activity of CaO prepared from different precursors(optimal temperature,60 min).

Fig.12.Effect of different additive content on hydration activity of CS-CaO at 900°C for 60 min(C:graphite,CC:CaCO3).
Reducing sintering of CaO is the most effective way to improve VF200 and decrease APD.Fig.15 shows the pore structure of CaO from CS with 5%graphite and 5%CaCO3.The APD of CS-CaO doped with 5%graphite and 5%CaCO3was shifted to 737.5nm and 427.9nm,respectively.The APD of CS-CaO was significantly reduced,indicating that graphite and CaCO3played a vital role in refining the pore structure of the CaO particles and accordingly increase the VF200.The VF200 of CS-CaO with 5%graphite and 5%CaCO3increases from 1.27%to 6.62%and 21.82%,as shown in Table 5.As a result,the hydration activity of the CS-CaO was enhanced,further confirming that the hydration activity was negatively affected by the increase of the APD and the decrease of VF200.

Fig.13.XRD pattern of CaO from CS(a)with 5%graphite or CaCO3,(b)with 10%graphite or CaCO3(900°C,60 min).

Fig.14.TG/DTG analysis of CS and modified CS under air atmosphere.
The mechanism for improved hydration activity by graphite and CaCO3was described in Fig.16(a)and(b).It is worth noting that hydration activity of CaO from CS added with 5%CaCO3is better than that of CaO from CS added with 5%graphite.Compared with adding graphite,the addition of CaCO3resulted in the increase in CaO in CS-CaO,and thus the hydration activity of CaO from CS with CaCO3was higher than that of CaO from CS with graphite.
Fig.17 shows the morphology of CS-CaO with the addition of graphite or CaCO3.It can be clearly seen that the structure of CS-CaO without graphite or CaCO3had an obvious sintered layer.However,with graphite or CaCO3addition,the sintered layer changed into the network porous structure.In addition,many spheres appeared on the surface of the modified CaO particles.As shown in Fig.17(d)and(e),EDX results indicated that these round spheres were CaO,and these spheres of CaO inhibited the continued sintering of the particles.It was speculated that the addition of graphite or CaCO3has a good inhibiting effect on the sintering of CaO particles during hightemperature calcination.Graphite or CaCO3was distributed as a dispersant in carbide slag particles,interrupting the sintering of CaO and retaining abundant pore structures[18].Therefore,the hydration activity of CS-CaO was improved.

Fig.15.Pore diameter distribution of CaO from CS with 5% graphite and 5% CaCO3(C:graphite,CC:CaCO3).

Table 5 VF200 of CaO from CS added with 5%graphite and 5%CaCO3
4.Conclusions
In this work,a solid waste from chlor-alkali plant,carbide slag was proposed as precursor for high activity CaO preparation.Four kinds of Ca-containing precursors,including CH,CC,CS,and LS,were calcined at different temperatures to investigate the relation between microstructures and hydration activity of CaO.The hydrolysis kinetics results show that the hydrolysis kinetics model of CaO is D4.Sintering of CaO particles resulted in changes in crystallite grain,APD and VF200,and thus lowering the hydration activity of CaO.The hydration activity of CaO increased with increasing VF200 and decreased with increasing APD and is the least affected by crystallite grain.Under the invariable calcination temperature,the core-shell structure formed by the addition of graphite or CaCO3to CS effectively inhibits the sintering of CS-CaO and improves VF200.Results also showed that the hydration activity of CS-CaO increased from 22.79°C·min−1to 27.19°C·min−1and 29.27°C·min−1,when the carbide slag was added by 5%graphite or 5%CaCO3,respectively.Correspondingly,the addition of graphite or CaCO3during the CaO calcination/carbonization cycle would inhibit the sintering of CaO particles and increase the cycle efficiency of the CaObased sorbent.Therefore,it is presumed that this method can not only increase the hydration activity of CS-CaO but also increase the CO2absorption activity of CS-CaO.

Fig.16.Mechanism diagram of adding(a)graphite and(b)CaCO3to improve CS-CaO hydration activity.

Fig.17.SEM images of CaO prepared from(a)CS,(b)CS with 5%graphite,(c)CS with 5%CaCO3,(d)and(e)correspond to EDX analysis of(b)and(c),respectively.
Acknowledgements
We want to thank Dr.PanPan Zheng from China University of Mining Technology(Beijing)for TGA analysis.
杂志排行

Chinese Journal of Chemical Engineering的其它文章
- Scaling of the bubble/slug length of Taylor flow in a meandering microchannel☆
- Analyzing of mixing performance determination factors for the structure of radial multiple jets-in-crossflow☆
- Particle-resolved simulation of packed beds by non-body conforming locally refined orthogonal hexahedral mesh☆
- Visual study on the characteristics of liquid and droplet in a novel rotor-stator reactor☆
- Molecular dynamics simulation of supercritical CO2microemulsion with ionic liquid domains:Structures and properties☆
- Modeling bubble column reactor with the volume of fluid approach:Comparison of surface tension models☆