棉花U3和U6启动子在CRISPR/Cas9基因组编辑体系中的功能鉴定
2019-01-31藏旭阳代培红李继洋蒲艳顾爱星刘晓东
藏旭阳,代培红,李继洋,蒲艳,顾爱星,刘晓东
(新疆农业大学农学院/新疆农业大学农业生物技术重点实验室,乌鲁木齐830052)
棉花是我国重要的经济作物、纺织工业原料和国家战略物资,其相关产业是国民经济体系的重要组成部分。解放后我国棉花种植业经历了几十年的蓬勃成长,棉花的总产量及单位面积产量得到了成倍的增长。然而棉花生产依然面临很多问题,如新疆土地盐碱严重,干旱缺水,导致大量土地不能充分利用[1],另外干旱和盐碱胁迫也影响棉花农艺形状、产量和品质等[2];棉花枯、黄萎病大面积发生,对棉花生产造成严重危害[3-4]。棉花品质低、成本高,导致棉花大量积压、植棉效益低下。为解决这些问题就需要培育出抗病抗逆优质高产的棉花新品种,而创制出这些新品种最终需要棉花生物学创新作为理论基础,需要棉花功能基因组学来解析农艺性状调控的基因网络[5-6]。但是广泛种植的棉花是异源四倍体,其基因组大而复杂,棉花基因功能的研究存在巨大困难。而基因组编辑技术的诞生则为棉花基因功能的研究和棉花农艺性状的基因调控网络解析提供了强大的技术解决工具。不仅如此,基因组编辑技术还可以用来创造出更多优异的棉花育种材料[7-8]。
ZFN、TALEN和CRISPR/Cas9是 三大主 要基因组编辑技术,其中CRISPR/Cas9技术简单、高效、灵活、方便等优点而为广大科研工作者所接受[9]。U3或U6启动子具有明确的转录起始位点,是CRISPR/Cas9基因编辑技术体系的重要元件。目前前人已利用植物自身的U3或U6启动子,在拟南芥、玉米、棉花、水稻、烟草、大豆等物种中成功建立了CRISPR/Cas9基因组编辑技术体系[10-14]。金双侠等[15]建立的启动子诱捕系统中的GUS基因高频率、多模式和时空特异性表达为分离基因及其调节序列、开展功能基因组研究奠定了坚实的基础。
雷建峰等[16-18]从海岛棉品种新海16中克隆了2个GbU3启动子、5个GbU6启动子,并验证它们在棉花中具有转录活性,但是这些启动子能否在CRISPR/Cas9体系中实现对棉花内源基因的编辑功能,目前还不很清楚。李继洋等[19]利用雷建峰已验证具有活性的其中2个启动子建立棉花的CRISPR/Cas9基因编辑技术体系,在新海16胚性愈伤组织中成功实现基因的靶向编辑,编辑效率约为27%。
通常CRISPR/Cas9基因编辑体系一次只能编辑1个基因,而对于多基因控制的遗传表型解析就显得效率低下。为此Ma等[20]构建了由多个U3和U6启动子驱动sgDNA并串联的CRISPR/Cas9基因编辑体系,能在拟南芥和水稻中同时编辑多个基因,大大提高了多基因编辑的效率,但该载体系统需要多个不同的U3和U6启动子。
基于以上的研究,为了验证其他具有转录活性的U3、U6启动子是否也能实现高效的编辑效率,为此本研究对克隆的GbU3-2P、GbU6-7P进行功能鉴定,筛选出更多能用于棉花CRISPR/Cas9基因编辑技术体系的U3和U6启动子,为构建棉花CRISPR/Cas9基因多敲技术体系提供更多有效的U3和U6启动子。
1 材料与方法
1.1 实验材料
原生质体制备所用的供试材料为新海16,由新疆农业大学农学院实验室保存。限制性内切酶购于Thermo公司;纤维素酶和离析酶由IUZMI株式会社代理提供,T4DNA连接酶、Blunt Zero平端载体、Trans5α感受态细胞、Taq DNA聚合酶、RNase A、1kb Plus DNA Ladder等均购自于北京全式金生物技术公司;Phusion超保真DNA聚合酶购于北京NEB公司。测序及引物合成均由新疆昆泰锐生物技术有限责任公司完成。T-GbU3-2P-5F-2::GUS-sgRNA,T-GbU6-7P-5F-6::GUS-sgRNA载体由本实验室保存[17-18]。35S-Cas9-ter-SK质粒由中科院上海生理生态研究所提供[21]。
1.2 载体构建
1.2.1棉花GGB基因编辑靶位点的设计选择与靶向片段的制备。本研究选取的启动子为GbU3-2P、GbU6-7P。GGB靶基因序列(sgRNA)如表1。靶序列设计在PAM(5'-NGG-3')(the protospacer-adjacent motif)位点前,长度为24个碱基(bp),并在PAM位点5'上游的切割位点处为限制性核酸内切酶识别位点。退火反应体系为20μL,上下游靶序列各10μL(表1);复性程序为:起始温度95℃,每5 s降低0.5℃,直至20℃,形成双链靶序列。
最后该靶序列经退火复性插入到经BbsⅠ酶切的T-GbU3-2P-5F-2::GUS-sgRNA,T-GbU6-7P-5F-6::GUS-sgRNA载体上(图1)。
1.2.2基因编辑载体的构建。将实验室之前构建好的T-GbU3-2P-5F-2,T-GbU6-7P-5F-6载体[17]保存菌液,摇菌(200 r·min-1,37℃,12~14 h)进行提取质粒。
对T-GbU3-2P-5F-2::GUS-sgRNA,T-GbU6-7P-5F-6::GUS-sgRNA载体进行酶切(BbsⅠ),电泳检测出现两条带(一条是GUS基因,条带长1 918 bp;一条是载体,条带长3 500 bp左右),回收目的片段。将靶序列与回收片段用T4连接酶连接过夜,使靶序列连接到经BbsⅠ酶切的载体相应位置上,使靶序列替换GUS片段,将U3/U6-靶序列-sgRNA载体,转化Trans-5α感受态细胞,对重组质粒进行酶切(KpnⅠ,HindⅢ)回收(GbU3-2P::靶序列-sgRNA、GbU6-7P::靶序列-sgRNA)。同时对35s-Cas9-ter-SK质粒用KpnⅠ、HindⅢ酶切,并回收35s-Cas9-ter片段分别与目的片段用T4连接酶进行连接、转化后对重组质粒用HindⅢ和KpnⅠ进行酶切鉴定,鉴定正确的质粒命名为GbU3-2P::GGB-sgRNA-Cas9、GbU6-7P::GGB-sgRNA-Cas9。

表1 本研究使用的引物信息Table 1 Primers used in this study

图1 靶序列插入位置示意图Fig.1 Inserted position of target sequence
1.3 富集高浓度PCR片段
对带有靶序列的载体进行Polymerase chain reaction(PCR)扩增,以富集到高浓度CRISPR/Cas9基因编辑的核心片段。具体方法参见李继洋等文章[19],采用超保真TransStart FastPfu Fly扩增带靶基因序列的编辑载体和空载体的核心系统区域,扩增体系参照说明书推荐体系,扩增片段覆 盖 有GGB靶 序 列 的GbU3-2P::sgRNA、GbU6-7P::sgRNA及35S-Cas9-ter两个核心元件和没有GGB靶序列的对照空载体核心元件,琼脂糖凝胶电泳检测正确后采用酚氯仿抽提和酒精沉淀法对其产物加以简单纯化。
1.4 原生质体的制备与转化
参考拟南芥已经建立的原生质体分离方法[22],采用纤维素酶和离析酶双酶体系制备棉花叶片的原生质体,略有改动。
首先配制含有1.5 %纤维素酶、0.4 %离析酶、0.5 mol·L-1甘 露 醇、20 mmol·L-1KCl、20 mmol·L-1MES、0.85 mL ddH2O,55℃水 浴10 min,充分溶解后,室温放置冷却后,再加入0.1 mol·L-1CaCl2、0.1% BSA的混合酶液5 mL,经0.45μm纤维素微孔滤膜过滤待用。
其次取15 d左右幼苗(真叶刚展开)的新鲜健壮的的子叶7~8片。用蒸馏水清洗表面,在吸水纸上晾干待用。准备好刀片和镊子,将下表皮撕去,之后叶肉细胞将裸露出来,平展于培养皿中,再用刀片将撕去表皮的部分切割成细长条,用镊子轻轻将去表皮的叶面置于酶解液中,使叶片完全浸泡在酶解液中。放置在回旋摇床上酶解(50 r·min-1)10~14 h,直至子叶叶肉完全酶解,酶解液成绿色为止。
待原生质体酶解结束后,将酶解混合液慢慢混合均匀,取斜面枪头蘸取含原生质体酶解混合液滴在载玻片上,室温下静置3~5 min,放在显微镜下观察原生质体的形态,拍照并做记录。若形态符合试验要求,就用少量的W5溶液重悬后,用枪头轻轻吸取10μL原生质体混合液,把盖玻片(18 mm×18 mm)放置在血细胞计数板上,将原生质体悬浮液从侧面缓缓打入到载玻片上,计算出原生质体的个数,重复3次统计。
最后原生质体制备好后,配制W5溶液(4 mmol·L-1MES、125 mmol·L-1CaCl2、154 mmol·L-1NaCl、5 mmol·L-1 KCl)、MMG溶液(0.4 mol·L-1甘露醇、15 mmol·L-1MgCl2、4 mmol·L-1MES)和WI溶液(0.5 mol·L-1甘露醇、20 mmol·L-1KCl、4 mmol·L-1 MES),取10~15μg富集的PCR产物,参照拟南芥原生质体转化的方法[23-24]将富集产物转化至棉花原生质体,于28℃,50 r·min-1避光过夜孵育转化后的原生质体。
1.5 编辑效应的检测
过夜孵育转化后的原生质体经镜检拍照后,采用12 000 r·min-1离心1 min收集沉淀,弃去上清液后,将棉花原生质体用植物基因组DNA提取试剂盒提取棉花原生质体基因组DNA,方法参照Trans Easypure plant Genomic DNA Kit操作要求,将提取的DNA经过浓度检测后,取等质量提取的DNA,在靶序列上找酶切位点(GbGGB采用ScaⅠ酶切)进行过夜酶切和不酶切2种处理,分别以2种处理的原生质体DNA为模板进行PCR检测,PCR反应体系参照NEB公司Phusion超保真DNA聚合酶推荐反应体系,采用先酶切后PCR的方法对靶序列内突变位点进行检测[19],通过琼脂糖凝胶电泳出现亮带情况,进行回收,初步检测出突变的PCR产物,然后克隆、测序,采用DNAstar和DNAMAN软件进行序列比对分析,检测棉花GGB靶序列内是否有突变位点。根据GGB靶序列的测序结果,计算突变率。
1.6 靶基因内不同区域突变频率的计算
为了排除出现的碱基突变是PCR过程中引入随机突变的可能性,为此对酶切前的棉花基因组DNA进行PCR扩增,引物见表1(Test系列),对扩增产物进行克隆。随机挑选100个长势均一的单菌落测序,通过DNAstar软件比对测序结果,以40个碱基为一个计算单元,统计包括靶序列区及其前后各2个40 bp区域突变碱基数,并计算相应区域碱基突变的频率,利用制图软件绘制Test区域碱基突变频率折线图。
2 结果
2.1 编辑载体构建
采用酶切方式检测上述构建的载体,结果表明构建载体大小符合预期设计,构建带有靶序列的U3:sgDNA、U6:sgDNA(图2),连接的GbGGB靶序列的GbU3-2P、GbU6-7P不同启动子的Cas9的编辑载体均于构建成功(图3)。
图2 构建带有靶序列的U3:sgDNA、U6:sgDNAFig.2 Construction of U3:sgDNA and U6:sgDNA with target sequence

图3 GbU3-2P::GGB-sgRNA-Cas9、GbU6-7P::GGB-sgRNA-Cas9基因编辑载体的酶切鉴定Fig.3 Identification of GbU3-2P::GGB-sgRNACas9、GbU6-7P::GGB-sgRNA-Cas9 gene editing vectors using restriction enzyme digestion
2.2 原生质体制备与转化
体系优化获得的原生质体在显微镜下呈圆球状,形态饱满,符合去壁细胞形态,经计算其数量可达106mL-1以上,通过显微镜检测观察显示,仅极少原生质体呈破碎状态,证明使用该体系制备的原生质体可以满足本研究的后续试验要求。经转化并过夜孵育后的原生质体,有少量细胞成簇聚集现象,再次在显微镜下观察细胞活性状态,说明转化后的棉花原生质体状态良好(图4)。

图4 棉花原生质体转化前后形态图Fig.4 Morphological change of cotton protoplasts before and after transformation
2.3 CRISPR/Cas9介导的棉花GGB基因靶位点的编辑和突变检测
将构建好的基因编辑载体进行转化,对获得的原生质体基因组DNA进行不酶切和酶切两种处理,然后以此为模板进行PCR扩增,结果如图5(1)(2)所示,酶切前携带靶基因序列的载体转化样品和空白对照载体转化样品都能扩增出预期500 bp左右的条带,并且亮度相近。而经ScaⅠ酶切后携带靶基因序列的载体转化样品,由于靶区域限制性内切酶识别位点被编辑突变,限制性内切酶不能进行切割,维持了模板DNA的完整性,则PCR扩增出预期条带的亮度比用ScaⅠ酶切后的空白对照载体的亮。
本试验对酶切后PCR片段进行回收连接、克隆并送测序,通过测序结果比对发现,在棉花新海16 GGB内靶位点有个别碱基发生突变的现象。发生突变的位置位于PAM位点上游限制性内切酶位点区域的基因靶位点上,GbU3-2P、GbU6-7P启动子的CRISPR/Cas9基因编辑载体均出现不同类型的突变(图6),全部为碱基置换(包括转换和颠换两种类型)。
为了排除碱基突变是由于PCR过程中引入随机突变的可能性,为此对酶切前的转化原生质体的基因组DNA进行PCR扩增,扩增产物克隆,随机挑选100个阳性候选单克隆菌落测序,通过DNAstar软件比对测序结果,以40个碱基为一个计算单元,对包括靶序列区及其前后各2个40 bp区域碱基突变个数进行统计,并计算相应区域碱基突变频率,利用制图软件绘制Test区域碱基突变频率折线图。结果显示靶序列20个碱基区域内的碱基突变频率远远高于两侧相邻各2个40 bp区域内碱基的突变频率(图7)。
上述结果证明基于海岛棉GbU3-2P、GbU6-7P启动子的CRISPR/Cas9基因编辑载体系统均能棉花中实现靶向基因编辑的功能。

图5 酶切/PCR检测CRISPR/Cas9基因组编辑效应Fig.5 Digestion/PCR for editing effect of CRISPR/Cas9

图6 GbGGB-sg RNA靶位点编辑效应(碱基置换型)测序检测结果Fig.6 Sequencing of GbGGB-sgRNA target site that were edited(type of base substitution)
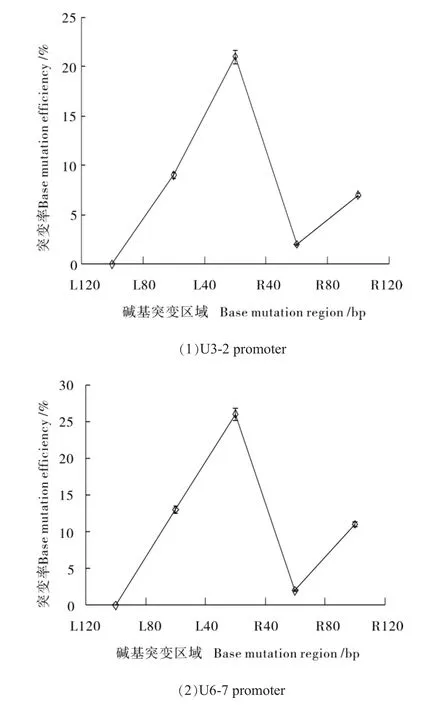
图7 Gb GGB-sg RNA2靶位点区域(guide RNA)及其两侧位点的突变效率分析Fig.7 Mutation efficiency of GbGGB-sg RNA2 target site(guide RNA)and its two flanking region
3 讨论
由RNA介导的CRISPR-Cas9基因编辑系统,作为一项全新的第三代人工核酸酶技术,其载体构建简单、成本消耗低,是作物功能基因组学和分子育种研究的强有力的工具,目前CRISPR/Cas9系统已经应用于多种植物的基因组编辑研究[25],该技术在植物中的研究已逐渐趋于成熟,而CRISPR/Cas9基因组编辑技术刚刚在陆地棉中成功建立[26]。但利用棉花内源U6或U3启动子在棉花中建立CRISPR/Cas9基因编辑系统的应用报道却很少。雷建峰已经从棉花中克隆了2个U3启动子、5个U6启动子,并验证它们在棉花中具有转录活性[18],但是基于这些启动子的CRISPR/Cas9系统能否在棉花中实现对内源基因的编辑,目前还不清楚。本研究利用已克隆的1个棉花内源的U3启动子和1个U6启动子构建了棉花的CRISPR/Cas9基因组编辑技术体系并成功地实现了基因编辑效应,为建立多敲CRISPR/Cas9基因组编辑技术体系提供了更多的启动子。
3.1 CRISPR/Cas9基因组编辑效应检测
采用先酶切后PCR的方式是一种简单、快速、有效的检测基因编辑效应的方法。Chen等[11]利用CRISPR/Cas9基因编辑系统对陆地棉的两个基因(GhCLA1和GhVP)在棉花子叶原生质体中进行了定点突变研究,试验结果表明靶标位点大多数情况是单个碱基的替换,少数是单个碱基的删除或插入。本研究利用已克隆的GbU3-2P、GbU6-1P、GbU6-7P启动子构建了棉花的CRISPR/Cas9基因组编辑技术体系并在新海16叶片原生质体中进行功能鉴定,结果显示靶位点的突变类型全部为碱基替换,未检测到碱基缺失和碱基插入,这与Chen等[11]的研究结果类似,但稳定转化能检测到较高频率的碱基缺失和碱基插入。可能的原因一是与稳定转化相比,原生质体瞬时转化的基因编辑系统的相关成分在原生质体中表达水平较低,且不能持续稳定表达,也就不能高效持续切割靶序列,而获得靶序列破坏严重的结果。第二方面的原因可能是选用的U3和U6启动子转录活性不高,不能高效产生SgRNA去引导Cas9高效持续切割靶序列。原因3可能是我们测序的克隆数依然偏少,没能检测到本来就比例偏少的插入和缺失克隆,进一步扩大测序的克隆数,可能就能检测到插入和缺失的突变类型。
空载体对照转化样品的模板DNA不会被编辑突变,限制性内切酶可以进行切割,理论上应该不会出现PCR扩增产物。然而我们空载体转化样品却出现了较弱的PCR条带,尽管我们增加了限制性内切酶量,也延长了酶切的时间,但对照依然有PCR产物。原因可能是棉花属多糖多酚植物,基因组在提取过程中残留的多糖和酚类物质在酶切反应时都可能抑制限制性内切酶活性,导致靶序列模板酶切不彻底,再经PCR扩增时,对照样品基因组DNA酶切后PCR产物依然还有微量的条带[27-28]。
PCR扩增常常会引入随机的碱基错配,即使使用高保真的DNA聚合酶,也会如此。本实验中靶序列单碱基的改变是否是因为PCR扩增过程中碱基随机配对错误产生的突变?为排除这个可能性,对靶位点及靶位点两侧的DNA序列突变的频率进行了测定,结果显示非靶序列位点处确实也可以检测到单碱基替换,但其突变的频率远低于靶位点碱基的突变频率,表明靶序列区碱基的突变确实是CRISPR/Cas9系统靶向编辑的结果。本研究结果表明棉花内源U3-2P、U6-7P启动子实现了CRISPR/Cas9介导的棉花基因组靶向编辑,克隆测序结果发现靶位点的突变多为碱基的替换,这与海岛棉、陆地棉原生质体中的基因编辑结果类似。
4 结论
本研究证明了棉花内源U3-2P、U6-7P启动子均能有效地驱动sgRNA的转录,实现CRISPR/Cas9介导的棉花基因组靶向编辑。该体系可以用于定向创制棉花的突变体,为棉花功能基因组学研究提供重要的技术工具。
