妊娠同族免疫性肝病-新生儿血色病1例报告并文献复习
2019-01-25牛会林陈晓文
陶 莉 牛会林 黄 蓉 赵 宁 陈晓文 魏 谋 周 伟
1 病例资料
男,G2P1,胎龄39周,出生体重3 260 g,因“产程延长”剖宫产娩出,前羊水清,后羊水Ⅲ度混浊,无窒息抢救史,Apgar评分1、5和10 min均为10分,胎盘、脐带情况不详。生后第2 d出现反应差、低血糖,以“新生儿感染、新生儿肺炎、新生儿气胸”收入当地医院,期间出现黄疸、肝功能异常:ALT 254.9 U·L-1,AST 421.1 U·L-1, ALB 18.9 g·L-1,TBIL 139.6 μmol·L-1,DBIL 43.8 μmol·L-1,凝血功能异常:PT >100 s,APTT >150 s,FIB <0.5 g·L-1,少尿,全身水肿(大量胸腔积液、腹水)、血小板50×109·L-1,予抗感染及对症支持治疗无效,反应仍差、黄疸渐加重、重度水肿,于生后11 d转入广州市妇女儿童医疗中心(我院)。
体格检查:体温38.1℃、体重4 550 g;反应差,哭声弱;肤色苍黄,皮肤可见散在紫癜,四肢水肿明显,前囟平软,口唇苍白;呼吸节律规则,双肺呼吸音粗,未闻及干、湿啰音;心率150·min-1,律齐,心音有力,心前区可闻及3/6级收缩期杂音;腹膨胀明显,腹壁皮肤发亮,静脉怒张,未扪及包块,肝、脾触诊不满意,肠鸣音正常,移动性浊音阳性;双侧阴囊水肿明显;肌张力正常;觅食、吸吮、握持、拥抱等反射减弱。
入院诊断:①反应差原因待查:新生儿败血症?遗传代谢性疾病?;②新生儿肝炎综合征;③腹水原因待查;④凝血功能障碍。
辅助检查:肝功能持续异常,TBIL、DBIL呈进行性升高,入院8 d达峰值,TBIL 252~359 μmol·L-1,DBIL 135.7~196.5 μmol·L-1,ALT、AST处于参考值范围。血氨81.8~264.5 μmol·L-1,凝血4项,PT 38.1~71.8 s、APTT 54.2~88 s、INR 3.56~7.28、FIB<0.45~2.61 g·L-1。PLT (18~74)×109·L-1)。腹部CT+增强显示,引流管留置下腹腔积液;门脉周围水肿;脾明显增大;胆囊周围积液、胆囊炎。胸腹水生化及常规检查未见异常,乳糜阴性。G-6-PD在参考值范围,直接抗人球蛋白试验(++)。总胆固醇 0.5 mmol·L-1,高密度胆固醇0.18 mmol·L-1, 低密度胆固醇0.53 mmol·L-1,载脂蛋白A1 0.13 mmol·L-1,载脂蛋白B 0.25 mmol·L-1。科萨奇病毒B组-IgM、巨细胞病毒-IgM、单纯疱疹-IgM、弓形虫-IgM、EB病毒-IgM、EB病毒-IgG、血和尿巨细胞DNA均阴性,多次血培养、胸腹水培养均阴性。血常规、PCT、真菌葡聚糖检测未见异常。骨穿:骨髓增生明显活跃,粒系、红系增生,巨核细胞可见,血小板少量散在或小堆分布。凝血因子Ⅱ 15.0%,凝血因子Ⅴ 16%,凝血因子Ⅶ 1.0%,凝血因子ⅩⅡ15%,凝血因子Ⅹ28%,凝血因子ⅩⅠ 11%,凝血因子ⅤⅢ 102%。尿有机酸分析:见大量4-羟基苯乳酸,中~大量4-羟基苯丙酮酸,中量4-羟基苯乙酸。血氨基酸+酰基肉碱分析:多种氨基酸升高;C0、C3含量偏低。NICCD(Citrin缺陷导致的新生儿肝内胆汁淤积症)基因阴性。心脏彩超示卵圆孔未闭。
入院后腹腔引流16 d,腹水减少。予头孢哌酮/舒巴坦、美洛西林钠抗感染;维生素K1、止血敏、血凝酶纠正凝血功能;还原型谷胱甘肽、腺苷蛋氨酸、熊去氧胆酸护肝;维持补糖速度5 mg·kg-1·min-1。因反复APTT、PT、FIB、血小板异常,多次输注血浆、冷沉淀、纤维蛋白原、血小板等效果差。入院32 d时患儿家属放弃治疗,患儿死亡。死亡诊断:急性肝功能衰竭,婴儿肝炎综合征
尸体解剖:肝脏8 cm ×6.5 cm×3.5 cm,肝重118 g(同体重婴儿平均肝重127 g),表面粗糙,细颗粒样,墨绿色,质地稍韧,边缘钝。镜下观察(HE染色)显示,肝脏(图1A,B):急性弥漫性肝坏死,胆汁淤积,灶性肝脂肪变性,肝细胞坏死、再生结节,脂肪变性,间质炎症浸润,肝细胞胞浆内见胆色素,小胆管内见胆栓,胆管增生;胰腺(图1C):分叶状排列,无明显坏死、变性,炎症浸润,胰腺腺滤泡胞浆见含铁血黄素颗粒。病理诊断:急性肝功能衰竭,胰腺铁沉着,含铁血黄素沉着?

图1患儿尸体解剖病理检查
注 A:肝细胞胞浆内见胆色素,小胆管内见胆栓,HE×200;B:肝细胞脂肪变性,HE×100;C:胰腺腺滤泡胞浆见含铁血黄素颗粒,HE×100
普鲁士蓝铁染色:采用普鲁士蓝-HE复染方法对患儿肝脏、胰腺、甲状腺、心肌及肺组织进行铁特殊染色,肝脏细胞胞浆内见可见铁颗粒沉积(图2A);胰腺腺泡上皮细胞内见较多蓝色铁颗粒沉积,(图2B);甲状腺部分腺泡上皮铁染色阳性;心肌及肺组织铁染色阴性。
膜复合攻击物C5b-9免疫组化:方法参考文献[2],一抗:小鼠单克隆 anti-C5b-9抗体,4℃过夜;二抗:辣根过氧化物酶(HRP)标记的羊抗鼠IgG抗体; ABC法显色;苏木精复染。以PBS为阴性对照。镜下观察,患儿肝细胞胞浆内可见较多棕黄色染色颗粒状物(图2C);对照组为胆道闭锁患儿,未见棕黄色染色颗粒。
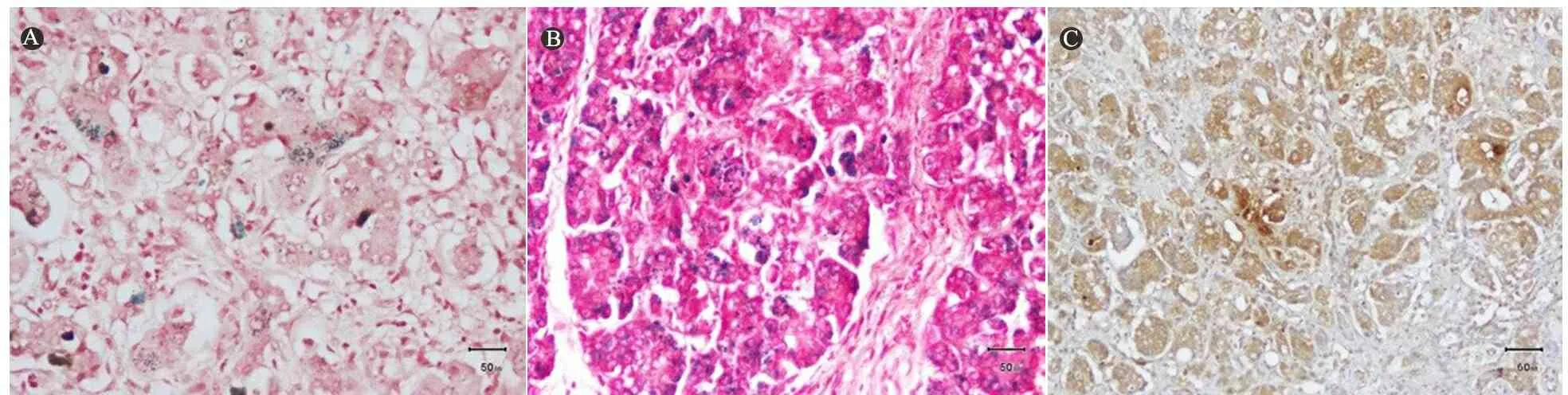
图2普鲁士蓝铁染色(×200)和膜复合攻击物C5b-9免疫组化
注 A:肝脏部分肝细胞胞浆内见蓝色铁颗粒沉积,B:胰腺腺泡上皮细胞内见较多铁颗粒沉积,C:膜复合攻击物C5b-9免疫组化肝组织可见较多棕黄色染色颗粒状物,×200
2 文献复习
以“neonatal liver failure[Title/Abstract]) OR neonatal hemochromatosis[Title/Abstract]) OR gestational alloimmune liver disease[Title/Abstract]”为检索式,检索PubMed和Web of Science数据库;以“新生儿肝衰竭”OR“新生儿血色病”OR“妊娠同族免疫性肝病”检索中国知网、维普和万方数据库,检索时间均从建库至2018年12月20日。共检索到255篇文献,均为英文文献。手工筛选到12篇文献[3-14],加上本文病例后共90例同族免疫性肝病(GALD)或新生儿血色病(NH)患儿,临床特点总结见表1。
90例中,胎儿7 例,新生儿83例;男21例,女22例,未描述性别47例;胎龄25~37周53例,37~41周37例;出生体重570~4 100 g,小于胎龄儿13例;胎儿水肿17例,羊水少21例,宫内生长发育迟缓33例,胎动减少5 例;生后表现铁代谢异常69例,肝功能衰竭62例,黄疸37 例,凝血异常58例,肝脾增大20例,腹水、水肿44例;NH确诊:肝外铁沉着阳性48例;MR胰腺铁沉着阳性19例,口腔黏膜活检铁染阳性 13例,病理胰腺铁染阳性 25 例;确诊GALD(肝活检或肝病理C5b-9免疫阳性)14例;铁螯合剂+抗氧化治疗36 例;IVIG+换血治疗20 例,单纯换血治疗9 例,单纯IVIG治疗4例,肝移植13例;死亡 55 例,存活35 例;随访 28例,随访年限10个月至10岁,预后均良好。
3 讨论
NH为一组临床综合征,表现为弥漫性肝脏病变及病理性多种肝外组织铁沉积,1961年Laurendcau等[1]首次描述了NH,但其病因学长期未明确。直到2010年Pan等[2]发现,NH属继发性病变,绝大多数由妊娠(胎儿期)GALD所致,此后陆续有散发病例报道[15]。NH是以新生儿期严重肝损伤甚至肝功能衰竭为显著临床特征的一组综合征,病死率极高。
NH为胎儿期严重肝损伤在新生儿期的延续性表现,大多数活产NH患儿有胎儿宫内异常史,如宫内发育迟缓、羊水过少或早产,为新生儿期最常见肝衰病因[15]。NH多于生后数小时,少数为生后数周出现肝损害表现,其临床表现以肝功能衰竭、多脏器衰竭、低血糖、凝血机制明显异常、低蛋白血症、伴或不伴腹水水肿(非免疫性水肿)、少尿为显著特点[16]。既往即使血培养阴性,临床极易误诊为重症败血症。患儿生后数天出现黄疸,绝大多数结合、非结合胆红素均升高,总胆红素常>300 mg·L-1。血清转氨酶水平与肝损程度不成比例,多数正常或更低,转氨酶升高者提示预后良好[16]。NH患儿还有甲胎蛋白(AFP)明显升高、转铁蛋白饱和度升高、血清转铁蛋白下降、血清铁蛋白升高等改变[16]。本文基于90例NH病例文献复习统计也显示,NH的临床表现以铁代谢异常(65例)最为显著,其次为肝功能衰竭(62例)、凝血异常(58例)、腹水水肿(44例)、黄疸(37例)、肝脾增大(20例)。 本文病例生后第2 d以低血糖表现起病,9 d内病情进展迅速,总胆红素和直接胆红素升高、凝血功能异常、少尿、全身水肿(大量胸腔积液、腹水)、血小板减少,血清转氨酶水平与病情严重程度不相符,呈现典型肝衰竭表现,符合NH临床特点。结合病史和相关辅助检查初步排除缺氧缺血性肝损害、细菌和宫内感染所致重症感染、遗传代谢性疾病。本文病例起初由于对NH认识不足,未进行铁代谢学及甲胎蛋白检测而缺乏相关临床资料。
NH主要病理表现为重度肝损,肝硬化明显、纤维化显著,主要集中于小叶内及中心静脉周围。残存或再生肝细胞为巨核形或假腺泡样改变,伴毛细胆管淤积,与成人急性或亚急性肝衰竭相似[15]。本文病例肝重118 g(同体重婴儿平均肝重127 g),光镜下表现符合急性弥漫性肝坏死病理改变。正常新生儿和许多新生儿期肝疾病均可出现肝铁沉着,因此,肝外而非肝铁沉着,为NH特征性改变,也是诊断NH必备条件[16]。NH肝外铁沉着最常受累包括胰腺管状上皮、心肌、甲状腺滤泡上皮、口咽和呼吸道黏膜层涎腺体,而胃肠腺体、甲状旁腺、脉络丛、胸腺、胰岛、腺垂体、透明软骨细胞较少受累[15-16]。本文病例普鲁士蓝铁染色结果显示,肝脏、胰腺、甲状腺有铁沉着,而心肌及肺无明显铁沉着。因此,结合肝脏病理改变及肝外脏器铁沉着,本文病例NH诊断明确。
曾认为NH为先天性铁代谢异常所致,被归入先天性家族性血色病(OMIM:231100)。此后数年,从临床特征判断NH为严重胎儿肝病的延续性表型。2010年Pan等[2]证实,绝大多数NH是由于妊娠(胎儿期)GALD所造成。GALD特异性靶向胎肝细胞,其抗原具有膜结合性特点,在胎儿期广泛表达,表达量数倍于成熟肝。此抗原可透过胎盘进入母体,一旦致敏,则母体内抗胎肝IgG形成,输入性结合于胎肝,产生损伤,其机制与胎儿固有免疫有关,终末补体级联效应通过经典途径被激活,形成膜攻击复合物,C5b-9免疫组化染色显示几乎所有病例均存在补体介导损伤,此为GALD的标志性特征[9]。本文病例C5b-9免疫组化显示肝细胞胞浆内较多棕黄色颗粒状物,因此,GALD诊断明确。曾有学者证实重症肝硬化新生儿,无肝外组织铁沉着(即非NH病例),其肝组织C5b-9阳性者而确诊GALD[17]。GALD作为胎儿肝病的明确病因,不仅明确了NH病因,还拓展了NH相关疾病谱,包括无肝外铁沉积的胎儿和新生儿肝病。
GALD-NH多需病理性诊断,未行病理检查者极易漏诊。对于新生儿重症肝病表现者,临床高度怀疑NH时,推荐口腔黏膜活检和胰腺MRI以确定有无肝外组织铁沉着[2,15-16]。无法确诊NH时(即肝外铁沉着阴性),即常规诊断手段无效时,可行肝组织C5b-9染色确定有无GALD[17]。
目前GALD-NH治疗措施有限,且疗效不尽人意。既往认为铁过负荷所致氧化损伤为主要病因,曾尝试抗氧化剂和铁螯合剂联合的鸡尾酒式疗法,但疗效
不佳,有效率仅10%~20%。基于对GALD-NH抗体介导免疫性损伤机制的了解,NH治疗策略渐转向免疫治疗,如双倍换血+大剂量IVIG(1g·kg-1)[16]。截止2012年,采用此疗法治疗了44例,35例存活(79%),4例接受肝移植(2例死亡,2例短期存活),5例未行移植死亡。因此,临床面对肝衰竭新生儿考虑GALD时,可给予换血+IVIG白治疗。虽然近年仍有肝移植治疗NH报道[11,18],但由于GALD-NH肝移植存在诸多问题,如时机选择、肝源稀缺、疾病本身自我修复性而不被推荐[16]。
NH母亲再孕子代发生NH风险仍较高,母妊娠期治疗可阻断再孕子代重症NH发生。目前推荐治疗:每次IVIG 1 g·kg-1,分别在孕14、16周,18周后则每周1次直至分娩。截止2008年,Whitington等[19]报道,接受产前治疗共110例NH母亲,1例因重症GALD在孕22周流产;2例分别在22、32周早产,均存活,预后良好;其余均无生长发育受限、胎儿肝病或胎儿窘迫表现,5例生后出现明显肝病表现。2016年Anastasio等研究结果同样获益[20]。这些数据说明孕期大剂量IVIG治疗可逆转再发性GALD,对胎儿及新生儿来说GALD已属非致死性疾病。
综上所述,GALD为胎儿期严重肝损害的主要病因之一,相关表型变异大,从几无临床症状到无肝外铁沉着的急性肝衰竭[7,10]。任何无法明确病因的晚期宫内胎儿死亡、新生儿期肝衰及肝硬化均应怀疑GALD。肝外组织存在铁沉着可确诊NH。目前认为GALD-NH为妊娠免疫性疾病。GALD孕母再次妊娠后子代再发风险仍极高,通过正确方法(包括活检)对NH胎儿或婴儿作出诊断是非常必要的。本例为国内首例GALD-NH报道,由于对其认识局限性,未能临床早期作出诊断。GALD-NH起源于妊娠期,且会出现严重不良妊娠结局,需要产科、新生儿、临床围产病理医师高度重视,积累更多资料,以能早期诊断、治疗,甚或妊娠期治疗,以避免重症NH出现。
