泊沙康唑(API)生产化工艺研究及杂质谱分析
2018-12-28郭彦飞包玉盛刘婷婷
郭彦飞,蓸 卫,包玉盛,刘婷婷
(南京海融医药科技股份有限公司,江苏南京 211100)
1 泊沙康唑简介
泊沙康唑主要适用于念珠菌属、隐球菌属真菌引起的真菌血症,呼吸、消化道、尿路真菌病,腹膜炎、脑膜炎等。临床上可用于曲霉病、接合菌病及镰刀菌病的治疗,亦可用于部分氟康唑耐药的念珠菌属感染的治疗。研究表明,泊沙康唑能广泛有效地治疗暗色丝孢霉病[1],提高皮炎外瓶霉感染者的生存率,作用呈剂量依赖性。泊沙康唑(posaconazole)是伊曲康唑的衍生物,最早2005年10月25日在欧洲首次上市的第二代三唑类抗真菌药物,美国的FDA于2006年上市,商品名为NOXAFIL,原研厂家为SCHERING,美国FDA于2009年9月15日又批准了泊沙康唑口服混悬液,于2013年11月25日批准缓释片剂,其缓释片剂型有两种剂型,分别为100mg缓释片和40mg/ml口服混悬液。又由2014年3月14日批准静脉注射剂,静脉剂型特别适用于需要静脉给药或者不能口服用药的患者。而且,部分患者可在初始治疗时选用Noxafil注射剂,之后逐渐转为口服剂型等。据介绍,Noxafil三种剂型均适用于免疫功能严重低下能够感染侵袭性曲霉菌和假丝酵母菌感染的高风险人群,比如:接受造血干细胞移植后,易患移植物抗宿主病的患者;因化疗导致中性粒细胞减少的恶性血液病患者。
泊沙康唑化学名称:4-[4-[4-[4-[[(3R,5R)-5-(2,4-二 氟苯基)-5-(1,2,4-三唑 -1-基甲基)氧杂戊环-3-基]甲氧基]苯基]哌嗪-1-基]苯基]-2-[(2S,3S)-2- 羟基戊 -3-基]-1,2,4-三唑 -3-酮;分子式:C37H42F2N804;相对分子质量:700.33;CAS登记号171228-49-2。
结构式:

2 路线筛选及工艺优化
文献[2-4]报道了泊沙康唑中间体的合成路线:(1)以2-(2,4-二氟苯基)丙烯醇为原料经Sharpless环氧化,亲核取代、缩合、还原等10步反应得到片段2。

(2)以2-羟基丙酸甲酯为原料,经亲核取代、水解、还原、加成等一系列反应合成化合物 a,以4-甲氧基苯基哌嗪盐酸盐和对氯硝基苯为原料,经偶联、还原、傅克反应、脱甲基化得b,再由a与b偶联得到化合物1。

在片段2的制备过程中用Sharpless环氧化反应生成环氧化物时,需使用手性试剂 L-DET(L-酒石酸 二乙酯)以及一些不稳定试剂,如(i-PrO)4Ti(四异丙醇钛)和 TBHP(叔丁基过氧化氢)。另外,此路线多个反应用 NaH做碱,危险大,片段2 共 计 10 步 反 应,现 有 文 献 收 率 (7.9%[5],6.7%[6],10.6%[7])周期长,收率低,不环保。片段1共计分为两个部分,也是共计10步反应,总文献收率也只在5%[8-10]左右,在片段1的合成过程中用到重金属及脱甲基试剂BBr3,高毒,环境不友好。从药物申报的角度考虑,在GMP车间不适合进行这种步骤长,周期长,环境不友好的工艺。因此我们将以片段1及片段2为起始原料,基于QBD的理念设计优化合成工艺,设计出适合于GMP车间进行大生产的工艺,同时根据当前的药物审评法规进行杂质谱的分析与研究
基于片段1与片段2为两个起始物料的合成路线如下:
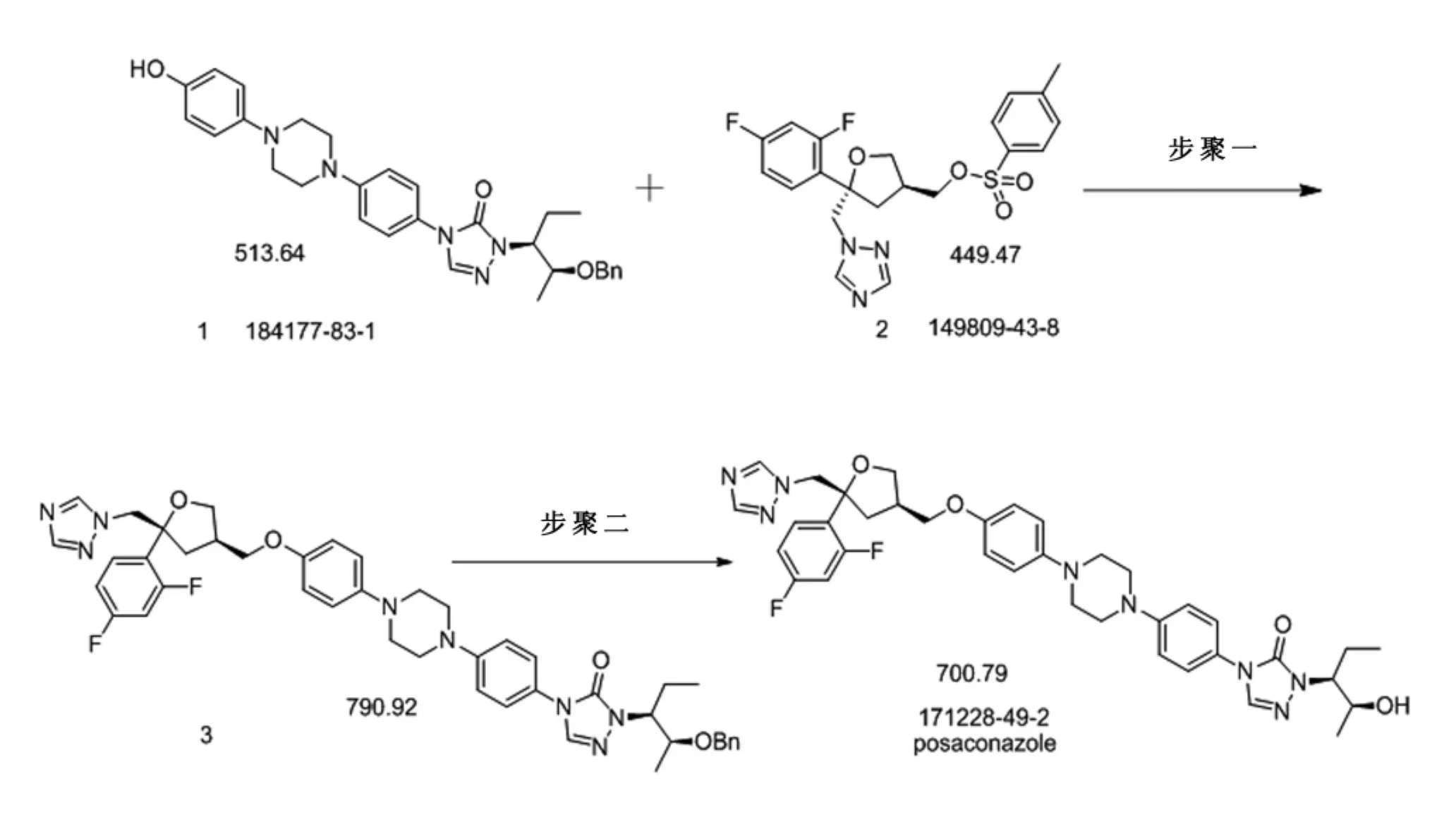
在步骤一的反应中,根据文献[4]报道的反应条件1 g的反应底物加入5倍量DMF为溶剂,然后再加入25%氢氧化钠水溶液在室温下反应30 h,在小试工艺研究中我们根据文献进行了复核,发现在室温下反应至48h,片段1始终反应不完,我们分别针对反应温度及氢氧化钠的量进行了小试研究,在此条件下原料始终反应不完。改用NaH做碱,DMF为溶剂,控温5~10℃搅拌30 h,可以反应完全,但是杂质谱明显比用氢氧化钠做碱要多,另外改用NaH做碱,DMF为溶剂,控温55℃ ±5℃搅拌10 h,可以反应完全,但杂质谱较杂,不经纯化直接进行下一步反应,不能得到单杂小于0.1%的产品。考虑到NaH在反应过程中会产生大量的氢气,同时NaH自身遇到潮湿的物品也会产生氢气,甚至发生爆炸,因此考虑到将来GMP车间的工业化生产,我们重新以氢氧化钠做碱进行工艺优化,同样得到了良好的反应效果,且中间体三只需一次结晶纯化,通过我们GMP车间的中试放大,证明工艺成熟稳定,具体工艺如下:
首先将1 kg氢氧化钠与纯化水加入配液桶中,搅拌均匀,配成25%的氢氧化钠溶液,降至室温,备用;在100L反应釜中加入DMSO(50L),开启搅拌加入片段1(5 kg),将配制好的氢氧化钠水溶液滴加入含有片段1的反应釜中,控温低于25℃,滴加完毕,控温30以下,慢慢加入片段2(4.5 kg),加料完毕控温40℃ ±5℃,搅拌反应15 h,TLC监控至片段1反应完全,反应终止,将反应液倒入200L反应釜中加入纯化水150L,室温析晶1h,过滤,纯化水淋洗三次;45℃ ±5℃,50L双锥真空干燥,得到中间体3,6.62kg,收率 86.4%,HPLC 纯度 97.46%,[M+H]+的 m/z为791.6,[M+Na]+的 m/z为 813.5(图1),核磁 δ(ppm,DMSO - d6):8.304 ~8.340(dd,2H),7.777(s,1H),7.46~7.489(m,2H),7.094 ~ 7.314(m,9H),6.931 ~ 7.012(m,3H) ,6.796 ~6.825(m,2H),4.514 ~ 4.590(m,3H),4.252 ~4.292(d,1H),3.979 ~4.058(m,2H),3.683 ~ 3.798(m,4H),3.166 ~3.326(m,9H),2.501 ~ 2.535(m,2H),2.148 ~ 2.174(m,1H),1.729 ~1.753(m,2H),1.214 ~ 1.235(d,3H),0.763~0.811(m,3H)(图2)。
在步骤二的反应中,根据文献 WO2013042138A2[11]和US2014343285A1(同族),在甲醇中加入中间体3,加入10%钯碳,在0.4~0.5MPa氢气压力下50℃反应4h,可以反应完全。另据文献[12]采用常压氢化在甲醇中加入中间体3,加入10%钯碳,通过加入酸来提高转化率,加入的酸有机酸如甲酸,无机酸如盐酸等,经小试研究,在不加压的情况不能够反应完全。氢化反应无论是高压氢化还是常压氢化都需要在氢化车间进行,还需要建立氢化车间,投入大,周期长,因此通过文献调研及工艺摸索寻求更优的脱苄基条件。根据文献[4],直接用氢溴酸做氢源及做溶剂,加热至50℃反应2~3h,可反应完全。经小试研究及不断的纯化摸索,发现始终有酸降解杂质无法控制在0.1%以下,不能够达到作为API的要求。HPLC图谱如图3所示。
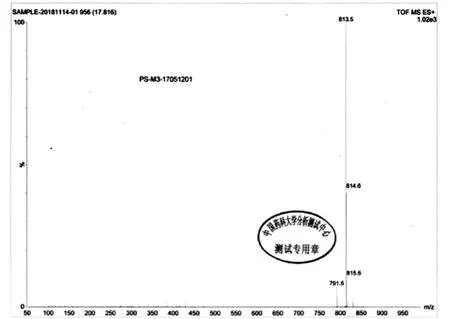
图1 泊沙康唑中间体质谱谱图

图2 泊沙康唑中间体核磁图谱
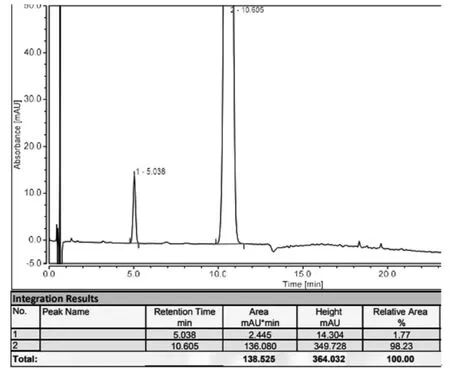
图3 HPLC图谱
因此考虑到将来GMP车间的工业化生产,我们在此研究的基础上改用盐酸作为氢源及溶剂,进行工艺优化,同样得到了良好的反应效果,通过我们GMP车间的中试放大,证明工艺成熟稳定,具体工艺如下:
在装配有尾气吸收装置的100L反应釜中,加入66L盐酸与6.6kg中间体3开启搅拌,开启加热装置,将反应液升温至40~45℃,保温反应7 h,TLC监控至反应完全,反应终止后,将反应液降温至10℃,加入二氯甲烷,将配置好的氢氧化钠水溶液滴加入反应瓶中,调节pH值8~10,滴加过程,控制反应液温度不高于20℃,滴加完毕,停止搅拌,静置20min分层,有机相备用,水相用二氯甲烷萃取两次(搅拌20min,静置20min),合并三次有机相,10%氢氧化钠洗涤一次,饱和氯化钠溶液洗涤两次,有机相减压浓缩真空(-0.08MPa)至液体无滴出,向蒸馏物中加入正己烷40L搅拌打浆30min,过滤,滤饼40~45℃,双锥真空(-0.09MPa)减压干燥6h,每2h,翻料一次。得到为泊沙康唑粗品5.41kg,收率为92.3%。
精制:在100 L反应釜中加入65kg甲醇,加入泊沙康唑粗品5.4kg,加热升温至55~60℃,全部溶解后,经0.5um滤膜导入洁净间100L结晶釜中,然后降温至0~5℃,搅拌析晶2h,离心,滤饼,双锥40~45℃,真空(-0.09MPa)减压干燥7h,得到白色泊沙康唑精制品4.52kg,收率83.7%。经质谱检测[M+H]+的 m/z为701.4,[M+Na]+的 m/z为 723.4(图4),核磁 δ(ppm,DMSO - d6):8.321 ~8.338(d,2H),7.777(s,1H),7.498~7.527(m,2H),7.233 ~ 7.309(m,2H),7.087 ~ 7.116(m,2H),6.927 ~ 7.012(m,3H),6.790 ~ 6.819(m,2H),4.573 ~4.656(m,3H),4.000 ~4.052(m,1H),3.674 ~ 3.805(m,5H),3.159 ~3.303(m,9H),2.365 ~ 2.432(m,1H),2.100 ~ 2.169(m,1H),1.683 ~1.729(m,2H),1.109 ~ 1.256(d,3H),0.719~0.767(m,3H)(图5)。
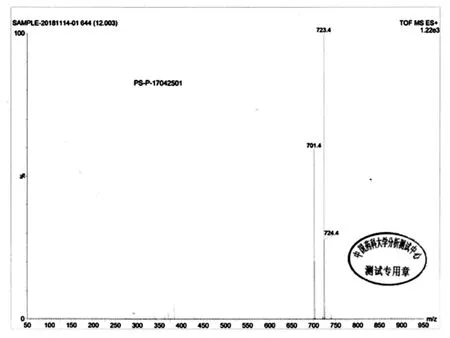
图4 泊沙康唑成品质谱谱图
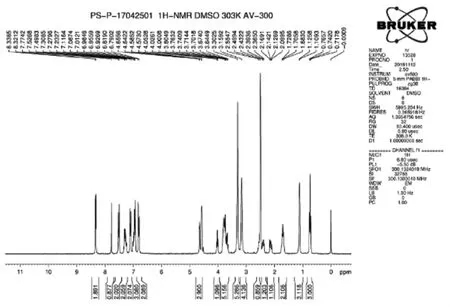
图5 泊沙康唑成品核磁图谱
目前泊沙康唑在步骤二中大部分企业还是采用钯碳氢化的方法,而采用钯作催化剂,不可避免引入重金属钯。2018年以来,欧盟、美国对元素杂质的要求越来越严格,中国在加入ICH之后对此项检测也会向国际靠拢。根据EMA对金属催化剂残留的相关规定及其解释,钯属于2B类金属,其限度为10ppm,必须采用ICP-MS法检测,目前大部分制药企业还没有ICP-MS,只能委托检验,时效上得不到保证。本工艺路线没有苛刻条件,操作安全,反应简单,后处理方便,完全可以在普通的原料药GMP车间进行生产。
3 杂质谱分析
泊沙康唑有四个手性中心,理论上有15个异构体(图6),所有异构体均是由片段1与片段2引入,合成难度较大,周期长,而且大大增加了泊沙康唑终产品方法学研究的工作量,因此我们需要从源头对杂质谱进行中控,通过理论分析、定向合成与色谱技术制备分离,可合成制备的杂质如图7所示。
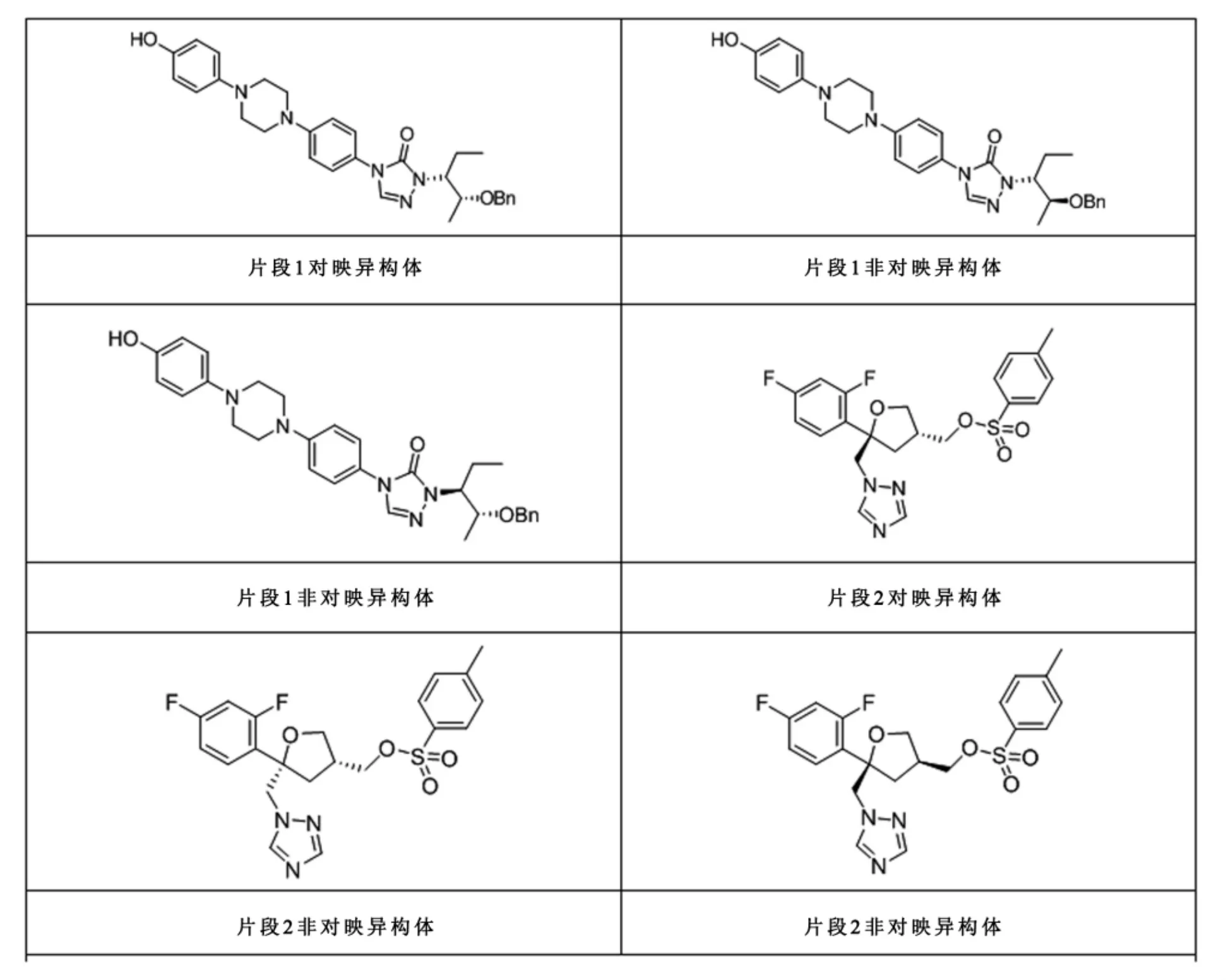
图6 片段1与片段2异构体
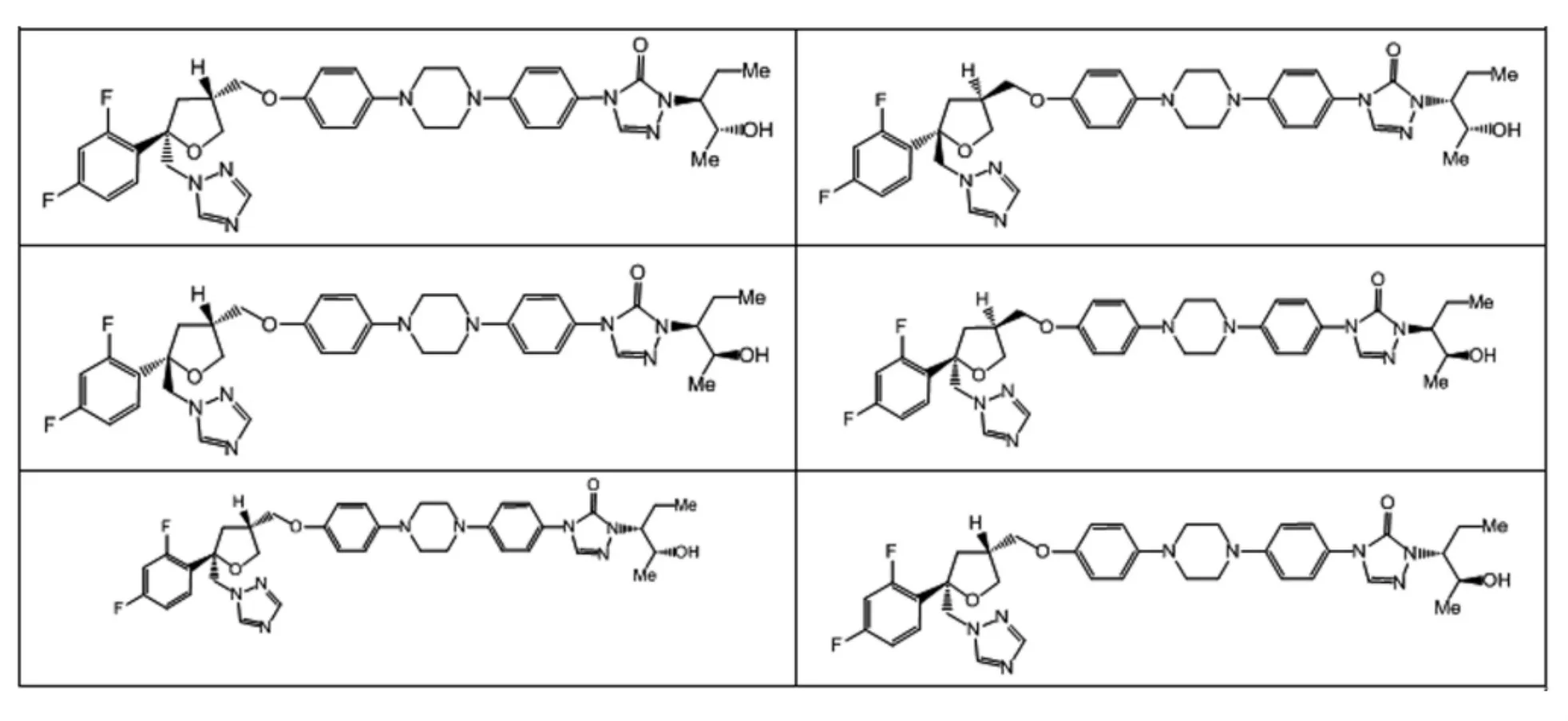
图7 泊沙康唑成品中通过定向合成及色谱技术可分离制备的异构体杂质
4 小结
通过定向合成及色谱分离技术,制备从起始原料至泊沙康唑成品的全系列杂质,并对分析方法进行了充分的摸索与方法学验证,分析指导合成工艺的开发,通过多批次的小试中试的工艺研究,此生产工艺合理有效,具有重现性和可靠性,能确保生产出来的产品质量符合要求,适合于原料药GMP车间放大生产。
