苏州不同果园土壤原核微生物的群落结构和多样性
2018-12-19陈宏伟胡翠英邱业先
钱 玮, 陈宏伟, 胡翠英, 邱业先
(苏州科技大学,江苏苏州 215009)
土壤微生物是农业生态系统中重要组成部分,主要包括细菌、真菌和古菌等,它们影响许多重要的生态过程,如植物对养分的获取,氮、碳、磷等物质的生物地球化学循环,并且对周围的土壤坏境十分敏感[1-2]。土壤微生物数量、群落组成及分布等指标已被公认为土壤生态系统变化的预警及敏感指标,目前土壤微生物多样性及功能的研究已引起国内外学者的极大关注[3]。
相关学者前期通过T-RFLP、PLFA、DGGE等手段分析了土壤细菌的多样性,并探讨施肥方式对细菌群落结构的影响[4-7]。但上述研究方法灵敏度有限,只能分析丰度大于1%的优势类群,而高通量测序技术可分析丰度大于1‰的类群,能较全面地反映微生物的群落结构,成为微生态学研究的主要方法[8]。
笔者运用高通量测序技术结合多元统计分析方法,分析了苏州地区果园土壤微生物群落结构以及微生物与土壤环境因子之间的关联,为研究不同土地利用方式对土壤生态系统的影响积累了有价值的试验资料,可为土壤生态系统的修复和健康状况评估提供理论依据。
1 材料与方法
1.1 材料与仪器
1.1.1 样品采集 取样地点位于江苏省苏州市金庭镇某农场(31°11.4′~31°11.6′ N,120°29.8′~120°30.2′ E),金庭镇位于中国第二大淡水湖——太湖之中,是我国淡水湖泊中最大的岛屿,该地属北亚热带湿润性季风气候类型,年平均温度约16 ℃,降水量在1 000~15 000 mm,加上太湖水体调节作用,成为国家级现代农业示范园区。农场内主要种植茶树、枇杷、柑橘、蔬菜等经济作物,于2015年11月在农场的茶树林、枇杷林、柑橘林、青菜地、野草地内分别设置10 m×10 m样方,在样方内5点取样,每份样品设置3个重复。采集0~10 cm的土壤,去除植物残体和石块,混合均匀后分成2份,置于无菌袋中带回实验室-20 ℃保存,1份用于测定理化指标,1份用于微生物多样性研究[9]。
1.1.2 试剂 PowerSoil DNA提取试剂盒,美国MoBio;PCR扩增试剂盒、DNA胶回收试剂盒、PCR引物等均购自上海生工生物工程有限公司。
1.1.3 仪器 高速冷冻离心机,GL-20G-Ⅱ,上海安亭生产;小型台式高速离心机,5417R,德国艾本德生产;凝胶成像分析仪,Universal Hood Ⅱ,美国伯乐生产;PCR扩增仪,5332,德国艾本德生产;恒温水浴锅,HH·S21-4-S,上海精宏生产;高压蒸汽灭菌锅,YXQ-LS-50SII,上海博讯生产。
1.2 试验方法
1.2.1 土壤样品的理化分析 土壤含水率采用烘干恒重法测定;pH值采用电极法测定;有机质采用灼烧法测定;速效钾采用醋酸铵浸提-原子吸收光度法测定;速效磷采用盐酸-硫酸浸提-钼蓝比色法测定;速效氮采用碱解蒸馏法测定;氨氮采用氯化钾浸提-纳氏试剂比色法测定[10]。
1.2.2 土壤总DNA提取、扩增和高通量测序 使用PowerSoil DNA提取试剂盒,按照操作手册提取土壤中总DNA,采用引物515F(5′- GTGYCAGCMGCCGCGGTA-3′ )和909R(5′- CCCCGYCAATTCMTTTRAGT-3′ )扩增原核生物16S rDNA V4-V5区[11-12]。25 μL PCR反应体系中包含0.25 μL ETaq酶 (5 U/μL),2.5 μL 10×PCR Buffer,2 μL dNTP Mixture(各2.5 mmol/L),1 μL模板DNA(20 ng/μL),1 μL 引物F(10 μmol/L),1 μL引物R(10 μmol/L),17.5 μL无菌蒸馏水。PCR反应条件为95 ℃ 5 min;95 ℃ 30 s,50 ℃ 30 s,72 ℃ 30 s,共27个循环;然后72 ℃ 延伸5 min。3份重复的PCR产物混匀纯化后构建DNA文库,然后由上海美吉生物医药科技有限公司利用Illumina公司Miseq 2×250 bp平台进行测序。
1.2.3 测序数据处理和统计分析 高通量测序结果通过QIIME(Quantitative Insights Into Microbial Ecology) pipeline[13]进行分析。原始数据使用FastQC(http://www.bioinformatics.babraham.ac.uk/projects/fastqc/)进行质检,去除低质量片段后的成对双末端序列根据重叠区融合成一条序列,根据barcode标签判断序列来源并区分样品。然后利用Usearch算法[14]在97%相似度水平[15]下进行OUT(operational taxonomic unit)聚类,每个OUT的代表序列依据silva数据库采用RDP classifier[16]按照界、门、纲、目、科、属、种分配分类单元。然后用QIIME软件计算各样品的α-多样性和β-多样性。典范对应分析(CCA)运用R软件Vegan程序包完成[17]。
本研究其他的数据分析使用SPSS 15.0完成,图形制作使用R 3.1.2软件(R-Development Core Team,2014)完成。
1.2.4 登陆号 本研究所涉及土壤样本16S rDNA序列已经提交至NCBI SRA数据库,登陆号分别为SUB1054781、SUB1054794、SUB1054825、SUB1054826、SUB1054827。
2 结果与分析
2.1 土壤理化性质
土壤样品的理化性质检测结果见表1,枇杷树、橘树土壤中有机质含量显著高于其他样本,枇杷树土壤中速效磷、速效钾显著高于其他土壤,分别为271.5、96.4 mg/kg,而茶树土壤中速效钾、速效氮分别为31.7、12.3 mg/kg,显著低于其他样本。橘树、茶树、枇杷树这3种木本植物根际土壤呈酸性,生长草本植物的土壤呈弱碱性。

表1 不同土壤样品理化性质比较
注:表中数据为平均值±标准误;同列数据后不同小写字母表示差异显著(P<0.05)。A、B、C、D、E分别表示橘树土、茶树土、枇杷树土、青菜土、草地土,表2、图1、图2、图3同。
2.2 土壤原核微生物群落多样性分析
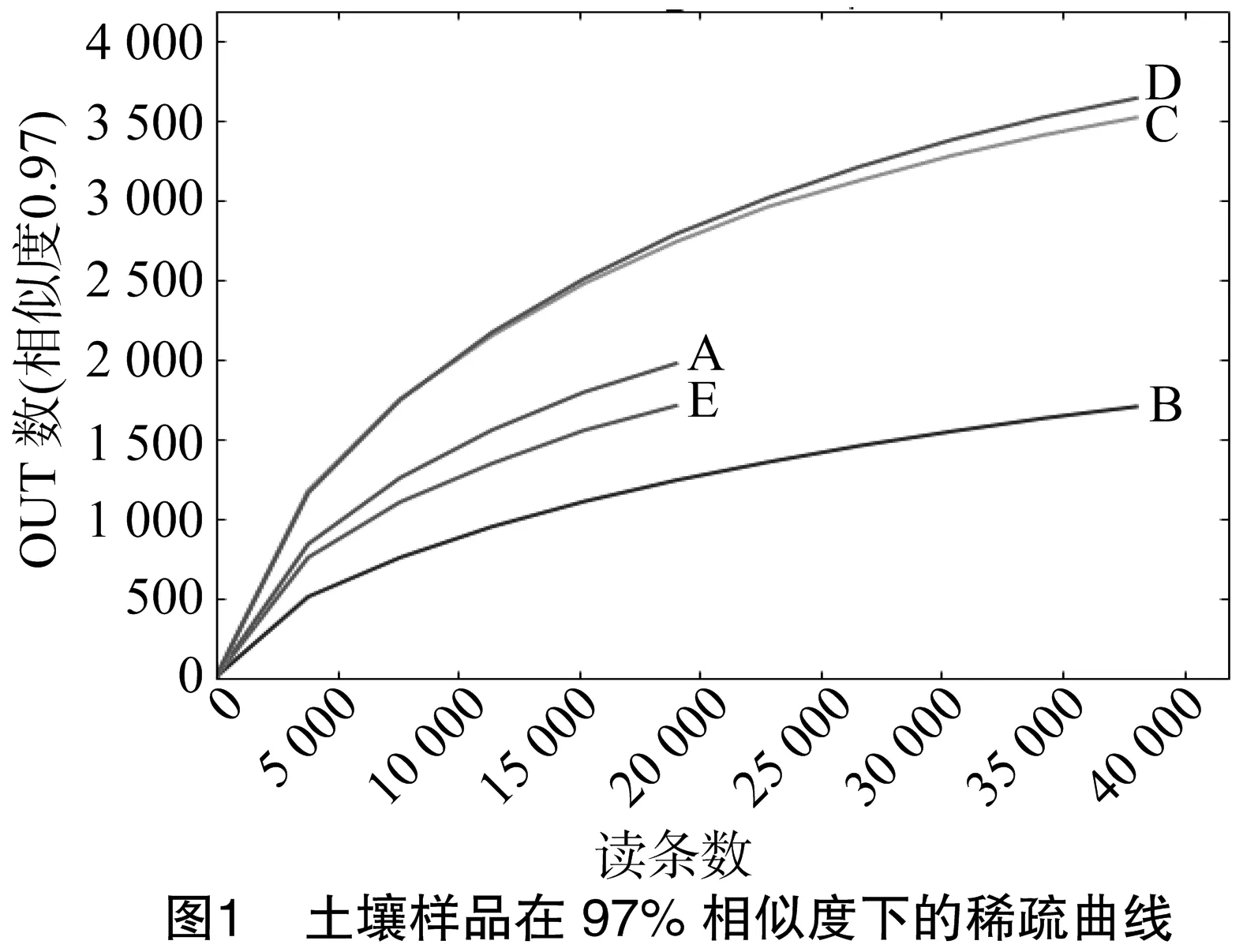
对5个样品进行高通量测序,共得到2.16 GB原始数据,经过质控、拼接和过滤处理后共获得179 831条有效的16S rDNA序列,序列长度分布在390~426 bp,平均长度为 396 bp,与扩增目的片段长度吻合。从图1可以看出,随着测序读条数的增加,曲线趋近于平坦,说明样本中大部分物种信息(OUT)已经得到,能够满足后续分析的需要,而继续增加测序数据量只会产生少量新的物种。为消除各样本间序列总数差异对后续分析的影响,把各样本序列数抽平到15 000条。然后在97%序列相似性水平下划分OUT,删除丰度小于总序列数0.01%的OUT,共获得960个OTU。5个土壤样品中平均含有570个OUT,其中青菜地土壤中原核微生物种类最多,为705个OUT;而茶树林土壤中微生物种类最少,为385个OUT。
不同样品中原核微生物群落的α-多样性分析结果见表2,由Chao指数和Ace指数可见枇杷树和青菜土壤样品中微生物种类最多,茶树和草地土壤中微生物种类较少。就Shannon指数而言,枇杷树土壤中微生物多样性最高,为 6.53;而茶树土壤中微生物多样性最低,为4.61;橘树、青菜和草地土壤的微生物多样性介于二者之间。

表2 不同土壤样品中原核微生物群落α-多样性指数
2.3 土壤原核微生物群落组成
从图2-A可以看出,苏州果园土壤中丰度较高的门为变形菌门、酸杆菌门、拟杆菌门、放线菌门、绿弯菌门,平均丰度分别为42.6%、22.0%、9.3%、7.2%、4.6%。其中枇杷树、青菜土壤中拟杆菌含量为16.9%,明显高于其他样品的 4.1%,但它们的酸杆菌含量为15.7%,低于其他土壤的 26.1%。草地土壤中属于古菌域的泉古菌门丰度为16.9%,远高于另外4个样品的1.5%。
从图2-B可以看出,土壤中丰度较高的科为黄色单胞菌科、酸杆菌科、Chitinophagaceae、红螺菌科、华杆菌科,平均丰度分别为10.4%、9.8%、9.3%、6.8%、6.6%。属于泉古菌门的SAGMA-X科微生物在草地样本中丰度为24.87,明显高于橘树和茶树样本的3.7%,而枇杷树、青菜土壤中SAGMA-X科属于低丰度微生物,平均丰度为0.03%。
选取平均丰度前100的OUT,根据分类信息和各OUT在不同样本中的分布,绘制土壤中原核微生物系统发生学分布图(图3)。从纲水平来看,土壤微生物主要分布于酸杆菌纲、拟杆菌纲、γ-变形菌纲和β-变形菌纲。从不同样本群落组成来看,枇杷树和青菜土壤中Cytophagaceae、黄杆菌属、厌氧绳菌纲和β-变形菌纲的丰度高于其他样本,但纤线杆菌纲、酸杆菌科、α-变形菌纲、γ-变形菌纲和泉古菌门微生物含量低于其他样本。


2.4 环境因子对群落结构的影响
为进一步分析环境因子对土壤微生物群落的影响,本研究在OUT水平进行了群落结构与土壤理化指标的典范对应分析(CCA)。从图4可以看出,第一轴对样本间物种差异解释度为66.1%,第二轴解释度为18.6%,总体解释度超过80%。,根据环境因子的轴长,可以判断pH值和速效氮含量对土壤微生物群落结构影响大于其他因子。速效氮与OTU5、6、9有较强的正相关性,与其他OUT呈负相关,而OTU5、6、9也是样本C、D的代表微生物类群。pH值与OTU2有较强的正相关,而与OTU8、1呈负相关,即酸性环境有助于提高OTU8和1在群落中的丰度,最终形成样本A、B类似的群落结构。这些主要OUT的代表序列提交至NCBI,BLAST比对结果见表3,表明多数OTU 代表序列与 GenBank收录序列相似度低于97%,可能为一些分类地位尚不明确的微生物,还需要进一步研究[18]。

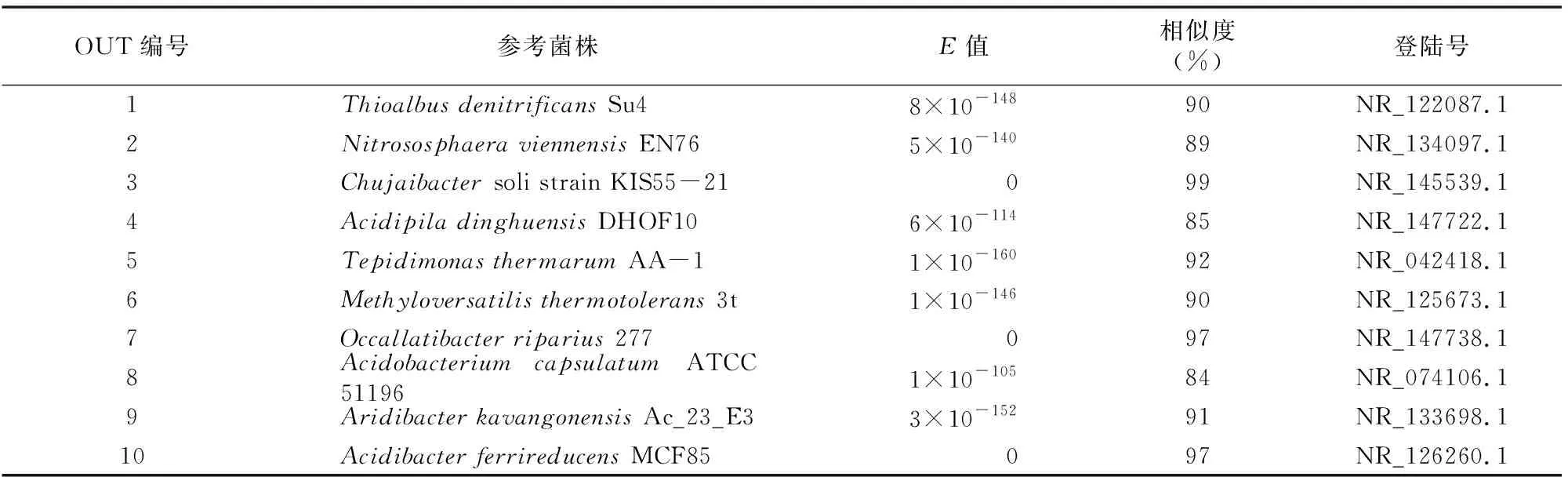
表3 主要OUT代表序列BLAST比对结果
3 讨论与结论
以宏基因组学方法探究了苏州果园不同土地利用方式对土壤微生物群落结构的影响,发现种植橘树和茶树的土壤微生物群落结构相似,种植枇杷树和青菜区域的群落组成基本一致,但是这些植物的生长都引起了土壤微生物群落的较大变化(相比草地而言)。最主要表现在于草地土壤中丰度较高的泉古菌门微生物在种植土壤中丰度大大降低,由16.9%降至1.5%,但是该类群微生物在土壤生态系统中的作用以及其丰度降低对生态系统的影响还需要进一步研究[18]。结合环境因子与群落结构的典范对应分析表明,土壤pH值和速效氮的含量是影响原核微生物群落组成的主要因子,但是这些环境因子的变化是种植方式(施肥或者松土等)不同引起的,还是由于植物和共生微生物协同作用引起的,目前仍是土壤微生物生态学研究的热点问题[19-20]。
