S-DABO类逆转录酶抑制剂的Topomer CoMFA及分子对接
2018-12-07仝建波秦尚尚
仝建波,王 洋,雷 珊,秦尚尚
(陕西科技大学 化学与化工学院 陕西省轻化工助剂重点实验室,陕西 西安 710021)
0 引言
获得性免疫缺陷综合症(AIDS),即艾滋病病毒,是引起人类免疫缺陷病毒(HIV)的一种致命疾病[1,2].HIV可分为I型(HIV-1)和II型(HIV-2)[3-5],两者之间存在一定的免疫交叉反应.据报道HIV-1是人类最容易传播和感染的病毒之一,所以绝大多数艾滋病都是由HIV-1而引起的[6].逆转录酶(RT)在HIV-1的复制循环中起着关键性的作用.因而,HIV-1逆转录酶一直以来都是抗艾滋病药物发展中主要的生物靶点[7].其中,非核苷类逆转录酶抑制剂(NNRTIs)和核苷类逆转录酶抑制剂(NRTIs)是如今HIV-1逆转酶的两大分类[8,9].NNRTIs作为高效抗逆转录病毒疗法(HAART)中重要的药物成分,由于作用靶点比较明确、结构多样化、高效毒性低、酶的结构清楚、与其他抗HIV药物具有协同作用等优点[10-12],所以在抗病毒药物的设计开发中意义重大,一直以来都是寻找新的抗艾滋病药物的重要倾向之一[13].
S-DABO属嘧啶酮类化合物,是最新已经发现的近50类NNRTIs中较具有代表性的一类,其化学结构是在C-6位上有取代苄基及C-2位上的烃(环烃)硫基对抗HIV活性有十分重要的影响[14].在基于配体的计算机辅助药物设计中三维定量构效关系(3D-QSAR)历来都扮演着重要的角色.其中CoMFA[15]与CoMSIA[16]是两种最传统且最典型的方法.本实验采用新一代CoMFA-Topomer CoMFA[17,18]对21个6-(1-萘甲基)取代S-DABO类化合物进行3D-QSAR研究.通过Topomer search[19,20]技术在ZINC分子数据库中进行虚拟筛选研究,然后选取了1个R1基团和8个R2
基团得到了8个活性不低于模板分子的新药物分子.最后采用分子对接技术研究了新设计的药物小分子配体与生物大分子蛋白(1RT2)之间的相互作用与结合方式,为实验工作者合成该系列新药物提供了理论指导.
1 方法与步骤
1.1 数据集来源与分子结构构建
本实验从文献[21]搜寻了21个具有确定结构的6-(1-萘甲基)取代S-DABO类逆转录酶抑制剂(见表1),其化学结构见图1,活性标度为IC50的负对数(pIC50),其中IC50为HIV-1病毒感染细胞半数抑制浓度.本文借助SYBYL2.0-X软件包中的Sketch Molecule模块来绘制分子的结构,采用最陡梯度下降法对所有分子进行能量优化,最终得到最低的能量构象.整个优化过程涉及到Powell能量梯度法,Tripos立场[22]等.其中最大迭代系数定义为1 000次,分子荷载电荷为Gasteiger-Huckel电荷,能量收敛设置为0.005 kcal/mol,其余参数都是SYBYL 2.0-X的默认值[23].

图1 化合物结构骨架

表1 化合物的结构及活性数据
续表1
No.R1'R2'Exp.pIC50Pred.pIC50Residual15(4'-OCH3)Phi-Pr7.527.200.3216CH3i-Pr6.396.45-0.0617(4'-F)Phi-Pr7.107.22-0.1218(4'-Cl)Phi-Pr6.496.96-0.4719OCH3i-Pr5.355.48-0.1320*OCH2CH3i-Pr5.855.670.1821(3',5'-CH3)Phi-Pr6.626.580.04
*标记为测试集化合物
1.2 Topomer CoMFA建模与模型验证
用于建立模型的训练集(training set)分子和用于验证模型外部能力的测试集(test set)分子根据随机取样原则分别定义为16个和5个.接着以活性最高的15号分子为模板采用Topomer技术将该分子切割成三部分,切割方式如图2所示.接着软件会尽量保持同样的切割方式对其他分子进行自动识别,如果个别分子与模板分子结构差异较大软件无法识别则需手动完成切割,最后这些碎片的3D构象按照必然的经验规则进行调整,生成Topomer 模型[24].自变量为R1和R2片段周围的立体场和静电场参数.并以IC50的负对数(pIC50)作为因变量,采用偏最小二乘回归法(PLS)[25]建模生成3D-QSAR模型.模型的内部预测能力是由留一法交互验证[26]测验的.而外部预测能力是借助建模所选取的5个测试集分子来达到目的.

图2 21个化合物分子生物活性的实验值与预测值的线性回归图
1.3 虚拟筛选
Topomer search可以非常高效地优化先导化合物和搜查完整分子,是一种极其迅速的3D配体的虚拟筛选工具,且不需要考虑大分子受体作用.采用Topomer search技术可以从相应的分子数据库中筛选出R基团(R-groups).其原理是:采用Topomer相似性进行侧链、骨架及完整分子的筛选,通过阈值的R-groups进一步打分,用模型预测其活性的贡献. 本文应用Topomer search技术对ZINC(2012)[27]分子数据库中的Drug-like类(包含130 000个分子)进行R基团的虚拟筛选,来得到具备较高活性贡献的R1和R2基团.除了Topomer间距设定成185之外,剩余参数均保持SYBYL 2.0-X默认值不变.
1.4 分子对接
采用Surflex-dock研究21个6-(1-萘甲基)取代S-DABO类逆转录酶抑制剂及新设计的分子与1RT2(取自TNK-651/HIV-1RT复合物晶体结构)的作用模式和作用机制.经过一系列修饰后,S-DABO以配体形式生成活性位点,用原型分子(Protomol)体现蛋白的结合位点.本文在筹备小分子的过程中,将配体的输出构象设定成20个,其余参数均不变,同时选择原配体结构为参比分子,来进行新设计的药物小分子配体与大分子蛋白(1RT2)的对接与打分.这些表征函数有Total-Score(总的打分,打分越高越好,高于6的输出构象被判定为较好的输出构象);Crash(碰撞打分,越接近0越好);Polar(极性相互作用打分,越大越好);C-Score[28](综合的打分标准,满分是5,大于4说明对接结果与实验值相似度高);Similarity(相似性参比,此值介于0与1之间,值越大,表明分子构象越相似.一般而言,大于0.8就说明两个分子叠合的比较好).
2 结果与讨论
2.1 Topomer CoMFA建模结果与评价
Topomer CoMFA是CoMFA与Topomer的联合技术,没必要手动进行分子叠合,可自动完成分子叠合.本实验采用三种切割方式来对数据集分子进行Topomer切割,切割方式见表2所示.利用Topomer CoMFA得到了3个不尽相同的3D-QSAR模型.表2展现了3个模型的建模结果.从表2可以看出,3个模型的主成分数N分别为3,3和4,F检验值分别为44.418,41.212和31.631,其中交互验证系数(q2)均大于0.5,非交叉相关系数(r2)均不小于0.6且非常接近1,说明3个模型都得到了较为可靠的结果.综合考虑和分析这3个模型,模型1不仅具有较高的预测准确性,而且Topomer切割方式可以较好的保持S-DABO的结构,更有利于研究核心基团与R基团的关系.所以本实验选取模型1进行后续的分子设计与分子对接实验.图2为模型1的训练集与测试集的实验值与预测值的线性回归图.从图能够看出化合物均匀的散布在45 °线上下,进一步表明模型1的预测结果较优[29].

表2 Topomer CoMFA建模结果对比
2.2 模型的三维等势图
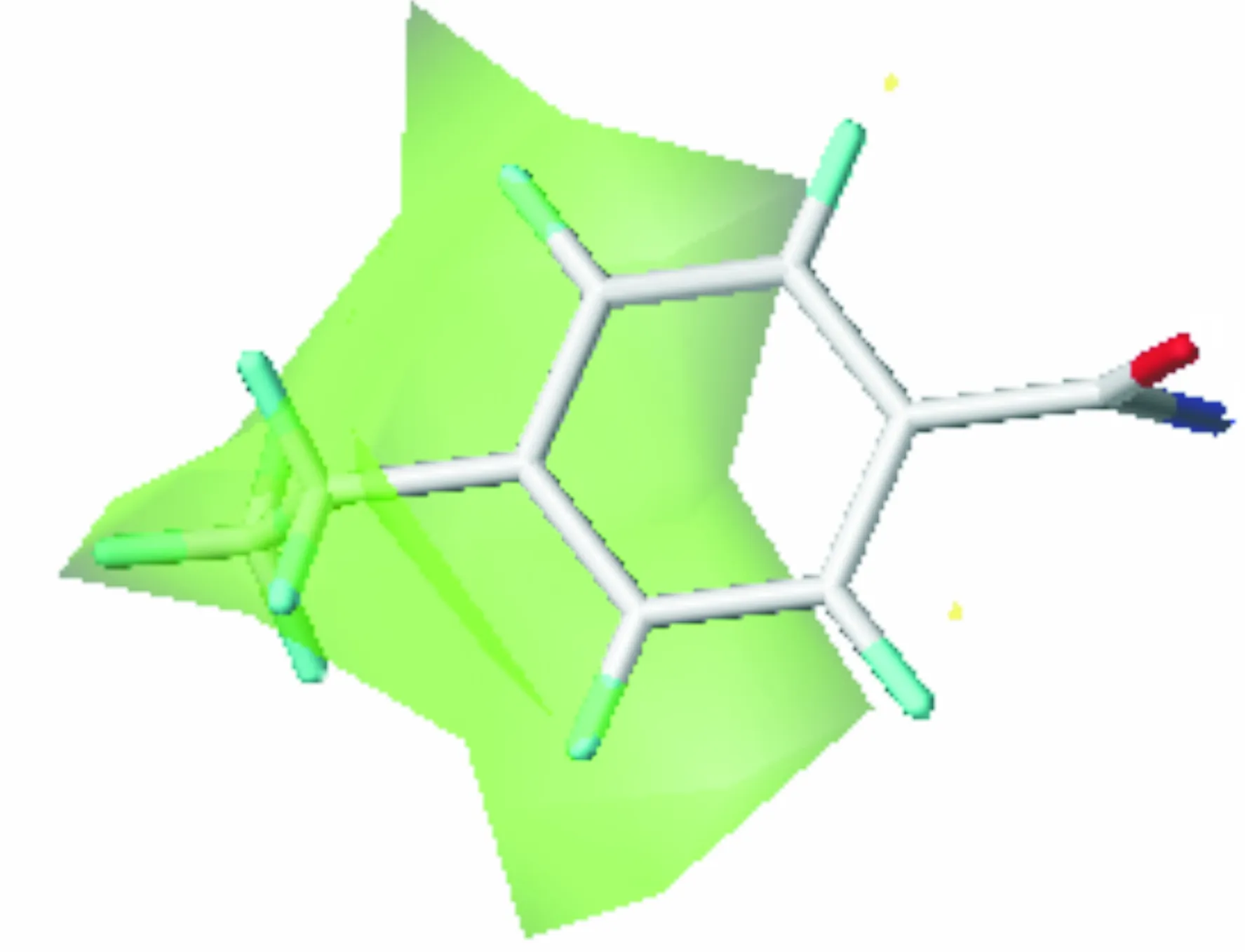
以活性最高的15号化合物为例,进行Topomer CoMFA三维等势图解析如图3所示.图3(a)和图3(c)分别表示R1和R2基团的立体场等势图,原则上,在立体场中,绿色区域表示引入体积较大的取代基对增大活性有帮助,黄色区域表示引入体积较大的取代基对活性的增大无益;图3(b)与图3(d)分别表示R1和R2基团的静电场等势图,红色区域表示引入负电性取代基对增大活性有帮助,蓝色区域表示引入带负电性的取代基对活性的增大无益[30].
在图3(a)中,整个基团都被一大片绿色的等势域包围,且没有出现黄色的等势域,表明了在R1基团处引入体积较大的取代基对活性提高有帮助.例如,化合物1、8、14在R1′取代基处苯环的4'位以较小的基团甲基取代了15号分子苯环4′'位的甲氧基,它们的活性都明显降低;化合物13的R1′取代基处是苯基,相比于15号分子也有所下降;化合物3、6、7、12、16、19、20以烃基、烷氧基取代了15号分子的芳香基,其活性降低程度更大;图3(b)中,几乎全被红色的等势域包围,说明在R2处引入带负电荷的取代基对活性提高有帮助,例如,化合物6、7、12、19、20由于R1′取代基引入的都是带正电荷的烷氧基所以活性整体来说相对较小;化合物3、16活性稍有提高,是由于它们的R1′取代基是电负性大于烷氧基的甲基;其余的化合物由于R1′取代基上都有苯环,所以活性都相对较高;还有化合物4的活性明显高于化合物5、化合物10的活性明显高于化合物11、化合物17的活性明显高于化合物18,都是由于氟基的电负性高于氯基. 图3(c)中R2取代基全被大片绿色覆盖,说明此处引入体积较小的取代基对活性提高无益,例如,在保持R2基团不变的情况下比较活性,其中化合物1<化合物8<化合物14;化合物2<化合物9<化合物15;化合物4<化合物10;化合物5<化合物11;化合物6<化合物12<化合物19;化合物7<化合物20,这些都是由于烃基的体积随着碳链增长而增加的缘故.图3(d)为R2取代基的静电场等势域,可以发现此图周围既没有蓝色也没有红色,说明R2取代基不受静电场的影响.为了验证这一结论,观察发现R2取代基分别是甲基、乙基、异丙基,由于这些都是给电子集团,电负性极小,所以可以不用考虑电负性的影响.
(a)R1基团的立体场等势图

(b)R1基团的静电场等势图

(c)R2基团的立体场等势图

(d)R2基团的静电场等势图图3 立体场和静电场三维等势图
2.3 分子设计
基于Topomer CoMFA模型1,对R1和R2基团进行虚拟筛选研究,得到了具有高贡献的R1为1 250个,R2为65个.以15号分子的实验值为参照,选择了1个R1和8个R2基团进行新药物分子的设计.最后得到的8个分子的预测活性均不低于模板分子,其中表3为8个分子的结构与预测活性.

表3 新设计分子的结构及预测活性值
续表3

序号结构预测活性值057.64067.64077.61087.61
2.4 分子对接
分子对接是借助Surflex-dock模块来完成的,分子结构取自TNK-651/HIV-1RT复合物晶体结构(PDB代码:1RT2).对1RT2的预处理步骤有:加氢、加电荷、修复主链、修复侧链、末端处理,除去原有配体、除去水分子和其他残基.经过这些处理后出现原型分子生成图(如图4),此图显示的是配体小分子在活性位点空间内与大分子蛋白的相互作用模式,同时也可以看出该对接口袋为一条狭长的口袋.新设计的8个分子的优化方式同训练集分子,将构象的最大输出值设为20,经过对接实验后,共得到160个输出构象,其他参数都是SYBYL 2.0-X默认[31].
在进行对接实验之前,首先进行可行性验证,其方法是先将晶体结构中的共晶配体抽出,然后再次与晶体对接,并以原来的共晶配体为参照.如图5所示,共晶配体在晶体结构中的构象与配体对接后的构象几乎重叠,这表明该方法能够有效的用于分子对接结果.
接着将8个新设计的分子分别进行对接实验,在所有输出构象中挑选出Total score打分在6以上,Crash打分接近0,Polar打分较大且CScore打分均为5的构象为例来分析抑制剂与蛋白的作用机制,共得到6个满足要求的构象[32].现选择03、04、06、08来进行研究,图6展示的是它们的作用模式图.03号分子中,配体中sp3杂化的O原子与LYS103中的氢原子形成氢键,其距离为1.860 Å,配体中sp3杂化的H原子与LYS101中的氧原子形成氢键,其距离为2.059 Å.04号分子中,配体中sp3杂化的H原子与LYS101中的氧原子形成氢键,其距离为1.767 Å.06号分子中,配体中sp3杂化的O原子与LYS103中的氢原子形成氢键,其距离为1.813 Å,配体中sp3杂化的H原子与LYS101中的氧原子形成氢键,其距离为2.094 Å.08号分子中,配体中sp3杂化的O原子与TYR318中的氢原子形成氢键,其距离为2.655 Å,配体中sp3杂化的H原子与LYS101中的氧原子形成氢键,其距离为1.855 Å.结果表明,一般地,配体分子与LYS101、LYS103、TYR318残基具有氢键相互作用[33].

图4 原型分子生成图(灰色区域代表原型分子)
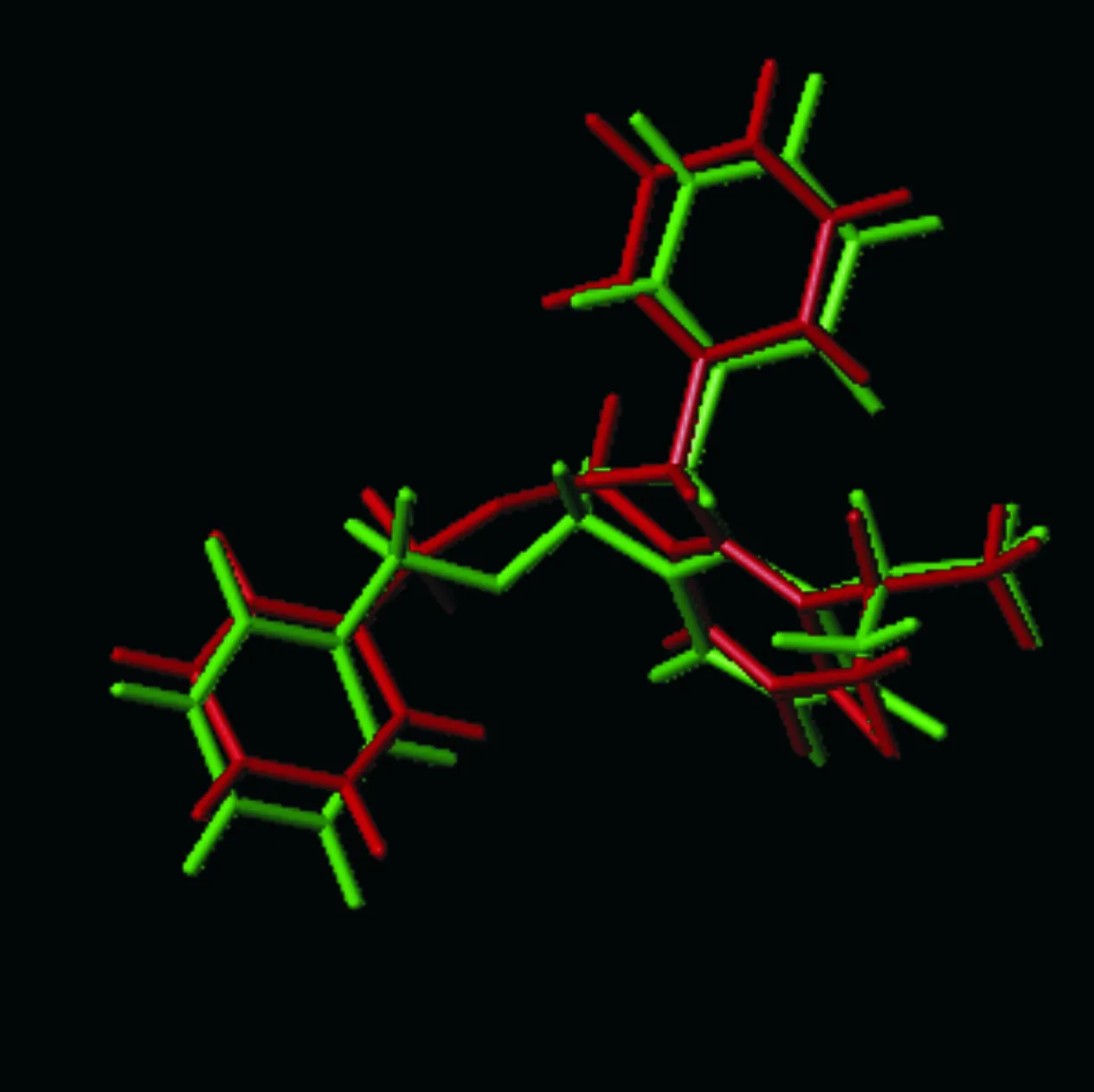
图5 晶体结构中共晶配体构象与对接构象叠合图(绿色棒状表示共晶配体对接构象,红色棒状表示共晶配体构象)

(a)03号分子与1RT2氢键作用
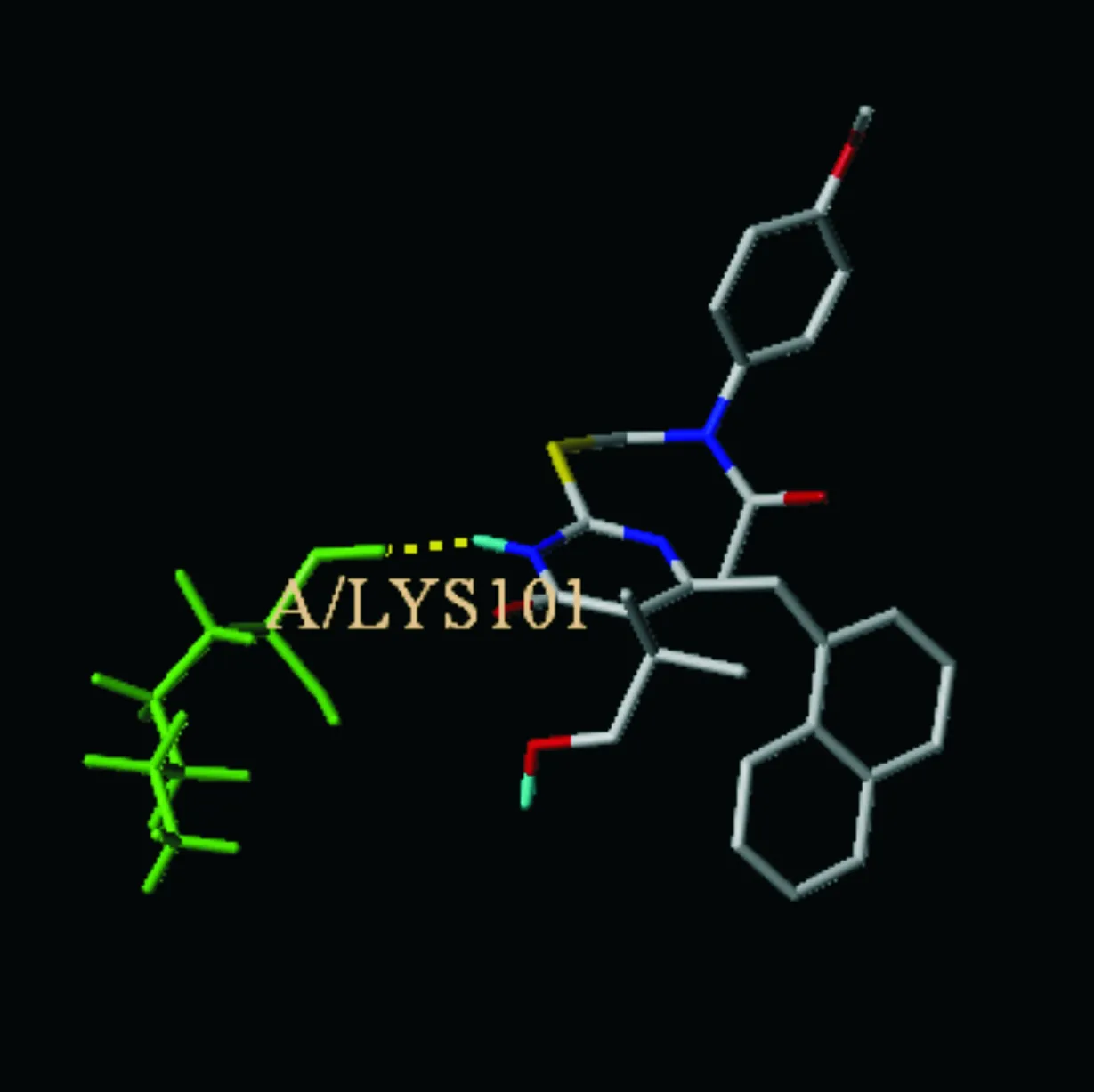
(b)04号分子与1RT2氢键作用

(c)06号分子与1RT2氢键作用

(d)08号分子与1RT2氢键作用图6 新设计的分子与1RT2氢键作用图(绿色棒状表示氨基酸残基,黄色虚线表示氢键)
为了进一步说明这个残基的重要性,对原数据集活性中活性最高的15号分子与活性最低的7号分子分别做分子对接.实验结果表明这两个分子的各项表征函数都符合要求.它们的氢键作用图见图7,其中7号分子中,配体中sp3杂化的O原子与LYS101中的氢原子形成氢键,其距离为2.183 Å,配体中sp3杂化的H原子与LYS101中的氧原子形成氢键,其距离为1.622 Å.15号分子中,配体中sp3杂化的H原子与LYS101中的氧原子形成氢键,其距离为1.851 Å.这些氢键作用使配体小分子与大分子蛋白结合更加稳定,同时也是药物分子抑制病毒的主要作用.

(a)7号分子与1RT2氢键作用

(b)15号分子与1RT2氢键作用图7 数据集分子与1RT2氢键作用(绿色棒状表示氨基酸残基,黄色虚线表示氢键)
3 结论
本实验利用Topomer CoMFA 方法对21个HIV-1逆转录酶抑制剂进行了3D-QSAR研究,最后得到稳定性及预测值均很好的3D-QSAR模型.以训练集化合物的结构片段为提问结构,然后采用虚拟筛选技术从ZINC数据库筛选得到有用的结构片段,并设计出8个新的具备较高活性的S-DABO类抑制剂.使得抗艾滋病药物分子的候选数据库得到了进一步扩充.运用Surflex-dock对抑制剂及其受体之间的相互作用模式进行了探索及探究,从而发现配体小分子中的某些原子与大分子蛋白中的一些主要氨基酸残基形成的氢键作用是药物分子发挥作用的主要依据.
