Synthesis of Mn O x-CeO2 Using Metal-Organic Framework as Sacrificial Template and Its Performance in the Toluene Catalytic Oxidation Reaction
2018-07-03LINXuetingFUMingliHEHuiWUJunliangCHENLiminYEDaiqiHUYunWANGYifanWENWilliam
LIN Xueting, FU Mingli,2,3,*, HE Hui, WU Junliang,2,3, CHEN Limin,2,3, YE Daiqi,2,3, HU Yun,2,3,WANG Yifan, WEN William
1 School of Environment and Energy, South China University of Technology, Guangzhou 510006, P. R. China.
2 Guangdong Provincial Key Laboratory of Atmospheric Environment and Pollution Control (SCUT), Guangzhou 510006, P. R.China.
3 National Engineering Laboratory for VOCs Pollution Control Technology and Equipment, Guangzhou 510006, P. R. China.
4 Centre for Clean Environment and Energy, Environmental Futures Centre, School of Environment, Griffith University, Gold Coast,QLD4222, Australia.
1 引言
挥发性有机物(VOCs)是细颗粒的关键前驱体,对大气环境和人体健康产生重大影响。催化氧化技术因其高效性和彻底性等优势已成为 VOCs治理的有效手段,广泛应用于众多行业领域中1–5。催化氧化技术的核心是催化剂,而锰铈复合氧化物催化体系又是其中的研究热点。锰铈复合氧化物一方面具备二氧化铈极强的储氧能力、负载金属时提供的较高的分散度及含有强酸强碱中心等特性6,7,一方面也具备氧化锰存在多种价态的特点,而具有良好的氧化还原特性的优点8,9,因而在VOCs的氧化中已得到广泛的关注10,11。然而传统的制备方法存在活性组分分散不均匀,形貌不可控以及颗粒团聚等问题,对锰铈复合氧化物的催化效果产生了一定的制约作用。因此本文选用新型金属有机骨架材料(MOFs)作为牺牲模板的制备方法,有望解决上述问题。
MOFs作为近十年来发展迅速的一种配位聚合物,是由金属中心离子和有机配体自组装形成的具有周期性网络结构的新型多孔功能材料,又称金属有机配位聚合物12。MOFs具有结构多样性、种类多样性和孔道尺寸大小可调控等特征,且囊括了主体金属中心离子和客体有机连接体具备的功能性特征,被广泛应用于气体储存、催化、传感和分离纯化等诸多领域,是一种很有前景的新型晶体材料。同时由于 MOFs金属离子的可调控性和配体的易裁剪性,MOFs被认为是制备特殊性质功能材料的理想牺牲模板,得到了人们的广泛关注13,相关研究工作已取得重要进展14,15。采用MOFs作为牺牲模板,可以得到具有可调控孔径大小,特定形貌以及高分散活性组分等特征的复合金属氧化物纳米颗粒。张宁16和徐力17等利用Cu和 Ce的 MOFs材料为前驱体制备活性组分高度分散的 CuO/CeO2催化剂并用于富氢体系 CO优先氧化,探讨了活性与金属活性物种分散度的关系以及影响分散度的相关因素。然而关于模板牺牲的过程中活性组分在载体表面的分布状态,以及此活性组分影响催化活性的本质机理还缺乏深入研究。
本文以铈基金属有机骨架材料作为牺牲模板,制备了MnOx-CeO2催化剂,并用于甲苯催化氧化。研究了不同锰含量的锰铈复合氧化物对甲苯的催化氧化性能,阐明了 MOFs模板牺牲的过程和活性组分在催化剂的分布状态,探讨了活性组分影响催化剂活性的机理。
2 实验部分
2.1 实验试剂
所需试剂: Ce(NO3)3·6H2O(硝酸铈六水合物,99.5%,阿拉丁科技有限公司)、1,3,5-H3BTC (均苯三甲酸,98%, 阿拉丁科技有限公司)、50% (w)Mn(NO3)2(硝酸猛溶液,阿拉丁科技有限公司); 所用的水均为去离子水。
2.2 模板和催化剂的制备
2.2.1 Ce-BTC模板的制备
称取 4.34 g (0.01 mol)的 Ce(NO3)3·6H2O 于小烧杯中,用50 mL去离子水溶解;另外称取2.10 g (0.01 mol)1,3,5-H3BTC于另一小烧杯中,用150 mL去离子水和200 mL乙醇溶解。混合上述两种溶液,在室温下搅拌2 min后,静置0.5 h。将混合液离心后所得的固体先后用乙醇(50 mL)和去离子水(50 mL)各洗涤3次,再真空抽滤。抽滤后的样品在120 °C下干燥12 h,制备出Ce-BTC模板。
2.2.2 MnO x-CeO2催化剂的制备
称取0.80 g的Ce-BTC于小烧杯中,加入20 mL乙醇和34 μL的50% Mn(NO3)2,混合均匀后室温放置 24 h,在 120 °C下烘干 12 h,最后在300 °C 下以 2 °C·min−1焙烧 6 h,即得到质量分数为1%MnOx-CeO2的催化剂。按上述相同的方法制备 3%MnOx-CeO2,5%MnOx-CeO2,8%MnOx-CeO2,10%MnOx-CeO2,催化剂标记为w MnOx-CeO2,(w指不同质量分数的金属Mn)。
2.3 模板和催化剂的表征
XRD测试在德国Bruker D8AdvanceX射线衍射仪上进行,光源为Cu靶Kα线,管电压40 kV,管电流 40 mA,步长 0.02°,扫描范围 2θ = 5°–80°。N2物理吸附-脱附测试在美国Micromeritics ASAP 2020型物理吸附仪上进行,样品预先在条件下150 °C真空脱气8 h。TG测试在德国NETZSCH STA449C型同步热分析仪上进行,样品质量为10 mg,升温速率为 10 °C·min−1,O2流量为 10 mL·min−1。EA 元素分析测试在美国 Thermo Scientific FLASH 2000上进行。ICP-OES在美国Varian 720上进行。SEM在日本Hitachi S4800/8010型扫描电子显微镜上进行。TEM在美国FEI f20透射电子显微镜上进行。H2-TPR测试在美国Micromeritics AutoChem化学吸附仪上进行,气体流量为 50 mL·min−1,气氛为 10% H2/Ar。测试前通He气在300 °C吹扫半小时,然后冷却至60 °C,开始测试,通入10% H2/Ar,吸附时间为60 min,测试区间设为 60–900 °C,10 °C·min−1的升温速率。XPS在美国Thermo Scientific EscaLab 250Xi型 X射线光电子能谱仪上进行。Raman测试在HORIBA LabRAM HR Evolution上进行,紫外光激发波长为325 nm,扫描范围在300–1350 cm−1之间,使用的可见激发光波长为532 nm,扫描范围在300–800 cm−1之间,使用CCD检测器作为拉曼信号检测器。UV-Vis紫外可见漫反射在岛津UV-2700型分光光度计上进行,扫描的波长范围是200–800 nm。
2.4 催化剂活性评价
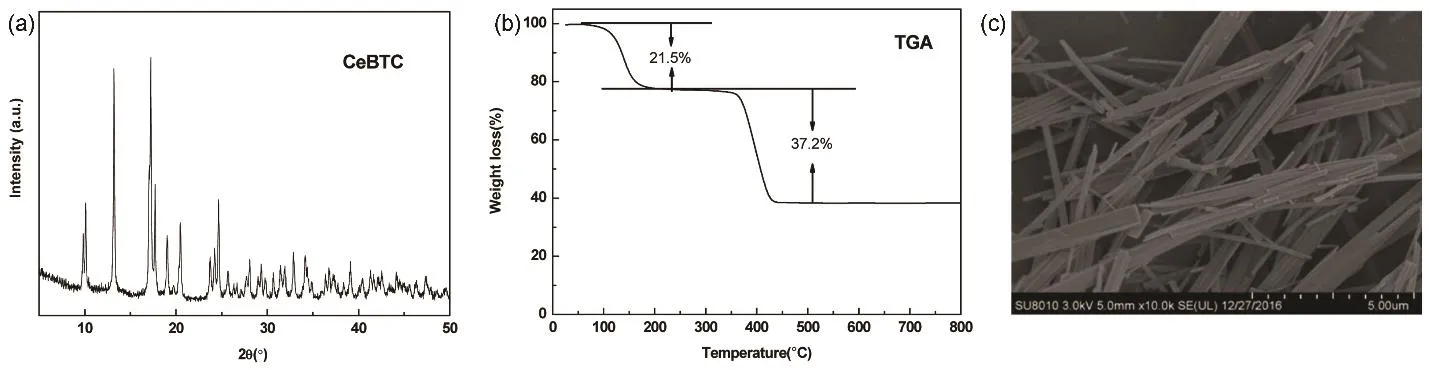
图1 Ce-BTC的(a) XRD,(b) TG和(c) SEM图Fig. 1 (a) XRD, (b) TG and (c) SEM of Ce-BTC.
甲苯催化氧化活性评价在内径为 8 mm的石英管反应器中进行。将200 mg 40–60目催化剂与800 mg石英砂混合均匀装入石英管内,反应气(4113 mg·m−3甲苯和干燥空气)气体流速为 160 mL·min−1,氧化反应热量由一个加热炉提供,反应温度由K型电热偶进行检测,由程序升温仪控制反应温度。反应气(重时空速 = 48000 mL·g−1·h−1)通过反应器前后都由一台气相色谱仪(GC-2014C,日本)进行检测分析,测量甲苯转化量和CO2生成量。甲苯转化率(Xtoluene)计算公式如下:Xtoluene=(Cin− Cout)/Cin× 100%,其中 Cin为反应气中甲苯进口浓度(10−6),Cout为甲苯出口浓度(10−6)。CO2产率(ωCO2)计算公式如下:ωCO2= (C(CO2)out−C(CO2)in)/[(C(toluene)in− C(toluene)out) × 7] × 100%,其中C(CO2)in为反应气中CO2进口浓度(10−6),C(CO2)out为 CO2出口浓度(10−6)。
3 结果与讨论
3.1 Ce-BTC模板表征
3.1.1 Ce-BTC模板的结构性质
对所合成的Ce-BTC进行XRD表征,结果如图1a所示,其主要特征衍射峰的峰位置和峰型均与文献18报道的相吻合,证实所制备出的Ce-BTC为纯晶体材料。Ce-BTC在30–800 °C范围内的热分解行为如图1b所示。其TG曲线分为三个阶段:第一阶段在30–170 °C,此阶段是分子孔道内吸附的水脱离孔道造成的质量损失;第二阶段为170–300 °C,几乎没有失重现象,说明MOF骨架在此温度维持稳定;第三阶段是 300–500 °C,此时晶体有大量的质量损失,归结为MOF材料骨架的分解和坍塌。Ce-BTC总计重量损失为58.7%,与理论计算中有机配体损失值59.6%相符,说明晶体分解基本完成。对Ce-BTC的形貌进行SEM表征,如下图1c所示,晶体呈现规则的棒状形貌,无杂质且晶型良好。
3.1.2 Ce-BTC模板焙烧温度的优化
Ce-BTC作为催化剂的模板,其焙烧温度的选择至关重要。既要保证MOF有机部分完全分解形成氧化物,又要避免温度过高引起晶粒团聚现象。根据上述热重的分析结果,在300–500 °C范围内选择了 5个温度点作为 Ce-BTC的焙烧温度,进而研究不同焙烧温度对模板的影响。
对 Ce-BTC模板焙烧后的材料进行 XRD表征,如图2a所示。通过观察,谱图的主要特征衍射峰均可对应于CeO2立方萤石结构。而且随着焙烧温度的升高,衍射峰峰强增大且峰宽变小,说明温度越高CeO2结晶度越好。此外,对其进行元素分析表征发现,CeO2-300 °C剩余的有机成分占比仅为1.30%,表明在300 °C下Ce-BTC模板中绝大部分的有机配体均分解完全。
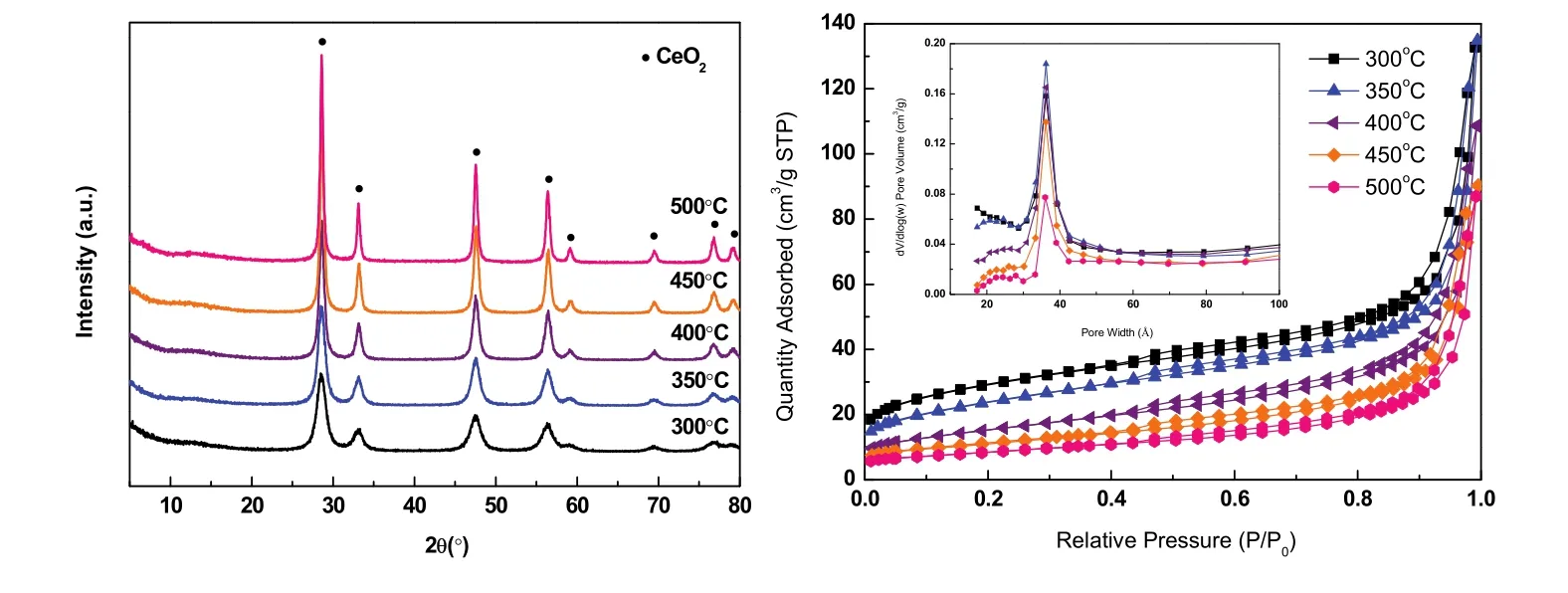
图2 Ce-BTC焙烧后CeO2的(a) XRD和(b)氮气吸附等温线图Fig. 2 (a) XRD and (b) N2 sorption isotherms of CeO2 calcinated from Ce-BTC.
各个温度焙烧后CeO2的N2物理吸附-脱附等温线如图2b所示。根据IUPAC分类,属于IV型吸附等温线。在低压部分(p/p0> 0.4),曲线出现回滞环,表明样品中存在介孔结构;从孔径分布曲线可观察到,样品孔径集中在 3.6 nm,且含有少量微孔,证明了样品中微孔与介孔并存。采用 BET方法计算得到 CeO2-300 °C的比表面积为 98.1 m2·g−1,且随着焙烧温度的升高,CeO2比表面积逐渐减小(见表1)。
为了更直观地观察CeO2的形貌与纳米结构,对其进行SEM和TEM表征,如图3所示。SEM表明CeO2-300 °C仍然保持Ce-BTC的棒状形貌,但是焙烧后表面粗糙且有空隙形成,归因于高温下有机配体分解从而在晶体结构内形成介孔结构。从TEM图可以看出,棒状CeO2是由无数纳米颗粒紧密接触而成,且可以清晰观察到内部孔隙空间,其纳米晶粒尺寸(4–6 nm)和 XRD 中Scherrer公式算出的(见表1)基本相符。
综上,Ce-MOF模板焙烧中发生:有机配体分解生成的CO2和H2O气体从晶体内部溢出,形成内部孔隙空间;而中心金属离子 Ce3+在空气作用下氧化成 CeO2纳米晶体颗粒。各项表征结果表明,在300 °C下Ce-BTC模板就能分解完全,形成具有棒状形貌、高度分散、高比表面积和纳米晶体颗粒等特征的CeO2晶体。因此,300 °C是Ce-BTC模板的最佳焙烧温度,后续实验中催化剂的制备均以此作为焙烧温度。
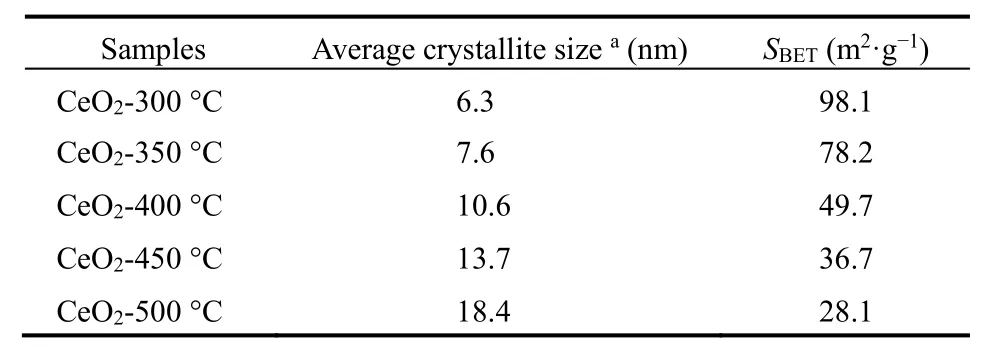
表1 Ce-BTC焙烧后CeO2的结构性质Table 1 Structure properties of CeO2 calcinated from Ce-BTC.

图 3 CeO2-300 °C的(a) SEM和(b) TEMFig. 3 (a) SEM and (b) TEM of CeO2-300 °C.
3.2 Mn O x-CeO2催化剂活性测试
以Ce-BTC作为模板,通过300 °C焙烧后制备的含不同 Mn质量分数的 w MnOx-CeO2催化剂用于甲苯的催化氧化反应。
其转化率随反应温度变化曲线如图4所示。从图中可以看出,随着Mn质量分数的增大,其转化率达到 100%所需的温度逐渐降低。在 100 °C时,均未显示出明显活性;当温度达到125 °C时,表现出一定的反应活性;在200 °C到275 °C的温度区间,反应活性迅速提高;而当达到300 °C,复合催化剂的甲苯转化率基本都达到100%。值得注意的是,当 Mn质量分数达到 5%、8%和 10%时,甲苯完全转化的温度分别为 252 °C、250 °C和258 °C,说明Mn含量超过一定程度时,MnOx-CeO2催化剂活性几乎保持不变。另外从图5可看出,不同Mn质量分数的复合催化剂对CO2的选择性也有一定差别。Mn质量分数为5%以上的催化剂在 275 °C后 CO2生成率逐渐稳定于 90%左右,说明对CO2选择性良好。下文将从催化剂体系的理化性质出发,对 Mn含量与活性之间的联系进行深入探讨。

图4 w MnO x-CeO2-300 °C的甲苯催化氧化活性Fig. 4 Catalytic performances of w MnO x-CeO2-300 °C for toluene oxidation.

图5 w MnO x-CeO2-300 °C的 CO2选择性Fig. 5 Selectivity towards CO2 of w MnO x-CeO2-300 °C.
3.3 Mn O x-CeO2催化剂表征
3.3.1 催化剂结构性质
图6是不同 Mn质量分数的 w MnOx-CeO2的XRD 图谱和 N2物理吸附-脱附等温线,表 2是w MnOx-CeO2的平均晶粒尺寸、晶格常数、比表面积和 Mn实际质量分数。由此可知,不同配比的MnOx-CeO2,从 1%到10% (测量的Mn实际质量分数与实验设计基本一致),均主要表现出CeO2的衍射峰,说明w MnOx-CeO2均表现出CeO2的(PDF No.24-0734)立方萤石结构。且可以观察到各配比的CeO2(111)晶面的衍射峰较纯CeO2发生宽化和钝化,其晶格常数也随着Mn含量的增加而减小,表明在引入Mn的过程中,Mn通过模板孔道进入以Ce金属簇为中心的MOFs框架内,并伴随着模板的焙烧进入CeO2的晶格结构形成Mn-Ce-O的固溶体。此时,离子半径较小的锰离子取代较大的铈离子进入晶格,使晶格收缩,其晶格常数减小11。另外,随着Mn含量的增加,w MnOx-CeO2的平均晶粒尺寸减小,表明 Mn的引入在一定程度上抑制了CeO2晶体的生长16,同时小晶粒的增多导致了比表面积和介孔数量增大。
值得注意的是,当Mn含量为5%及以下时,未观察到任何锰物种的衍射峰,表明锰物种以高度分散的形式存在于载体表面19;当 Mn质量分数达到 8%及以上时,XRD出现了 Mn3O4(PDF No.43-1002)衍射峰,表明此时催化剂表面形成了Mn3O4晶相。推测是Mn含量超过一定限值后,以氧化物晶态形式存在于催化剂表面。再结合 TPO活性分析,当Mn含量高于某一阈值的时候,这些以晶态形式存在的猛氧化物对活性无促进作用,说明此时体系物相结构发生了变化,出现了阈值效应20。于是认为,在引入Mn的过程中,除了小部分Mn进入CeO2晶格形成固溶体外,其余Mn主要分散在CeO2表面。当Mn含量低于单层分散阈值时,Mn3O4和CeO2表面某些活性位置相互作用形成表面化合物,单层分散于表面;当Mn含量高于单层分散阈值时,多余的 Mn以晶态形式存在于表面。由于Mn的单层分散和载体CeBTC的热分解同时进行,Mn组分分散到刚刚生成的、细小的CeO2颗粒表面,使其彼此隔开,起到了阻止颗粒烧结的作用20。这很好地解释了样品的晶粒尺寸随 Mn含量提高而减小,而比表面积及介孔数量随之增大的现象。因此下文使用了更多的表征手段对此进行印证。
3.3.2 SEM-HRTEM表征

图6 w MnO x-CeO2的(a) XRD和(b) 氮气吸附等温线图Fig. 6 (a) XRD and (b) N2 sorption isotherms of w MnO x-CeO2.
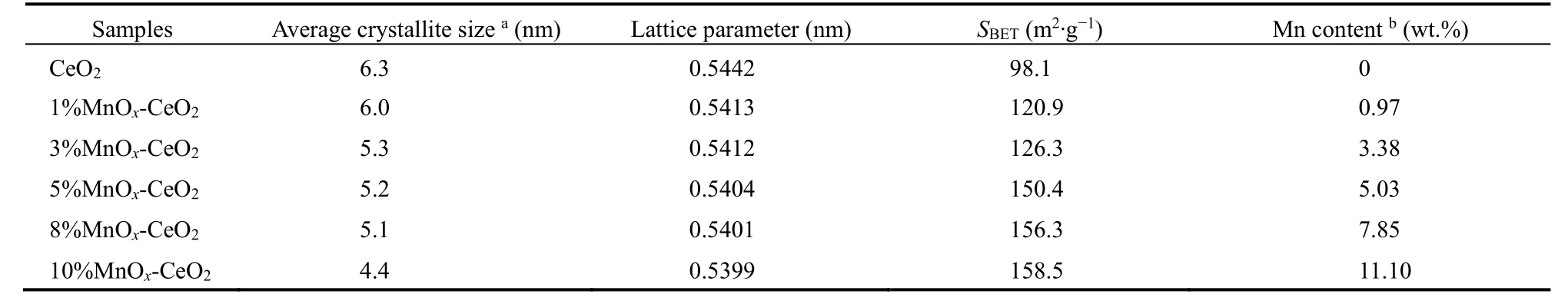
表2 w MnO x-CeO2的结构参数Table 2 Textural parameters of w MnO x-CeO2.
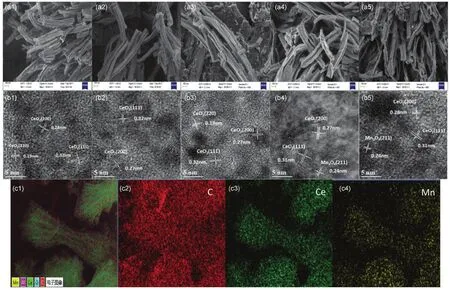
图7 (a,b) w MnO x-CeO2的SEM和HRTEM图;(c) 8%MnO x-CeO2的SEM-mapping图Fig. 7 (a, b) SEM and HRTEM of w MnO x-CeO2; (c) SEM-mapping of 8%MnO x-CeO2.
样品形貌表征和晶面构成如图7所示。从SEM图可以看出,在引入Mn之后部分催化剂断裂为碎片状,但总体仍然保持良好的棒状形貌,且在焙烧后棒状催化剂表面粗糙,有聚集的现象。观察不同配比催化剂的HRTEM图,发现Mn含量为1%、3%和5%的MnOx-CeO2主要暴露CeO2的(111)和(200)晶面,表明模板焙烧后形成的纳米晶体颗粒具有良好的结晶度;而8%和10%的催化剂除了暴露CeO2的晶面外,还能观察到Mn3O4的(211)晶面,与XRD结果相一致。通过EDS-mapping考察各元素在催化剂表面的分布(以8%MnOx-CeO2为例),发现Mn元素均匀分散在MnOx-CeO2表面。这些均印证了上文的猜想,即Mn在模板焙烧的过程中还以氧化锰的单层分散态和晶相形式分散在CeO2表面。
3.3.3 Raman表征
图8是w MnOx-CeO2催化剂的可见激发Raman谱图。其中,在465 cm−1处出现一个特征峰,归属为CeO2的立方萤石结构F2g的特征振动,而650 cm−1的特征峰被认为是MnOx中Mn―O―Mn伸缩振动峰,归属于Mn3O421。可以观察到,w MnOx-CeO2主要表现出CeO2的立方萤石结构,且相对于单一的CeO2均发生了一定程度的红移、宽化和钝化。这主要归因于CeO2结构中形成的缺陷和小晶粒尺寸。一方面Mn掺杂进CeO2的立方萤石结构诱导形成氧空位,致使晶格常数发生变化;另一方面与样品的晶粒尺寸引起的不均匀拉伸张力以及声子限制相关22,说明其拥有更小的晶粒尺寸,上述结果与XRD表征相一致。其中,Mn含量越接近5%,F2g峰的红移程度越大,说明此时Mn物种和CeO2表面存在强相互作用23。另外,值得注意的是,当Mn含量达到8%及以上时,观察到了Mn3O4的衍射峰,再一次证明了Mn在CeO2的成晶过程中,有一部分Mn以Mn3O4小晶粒形式均匀分散于CeO2表面24。
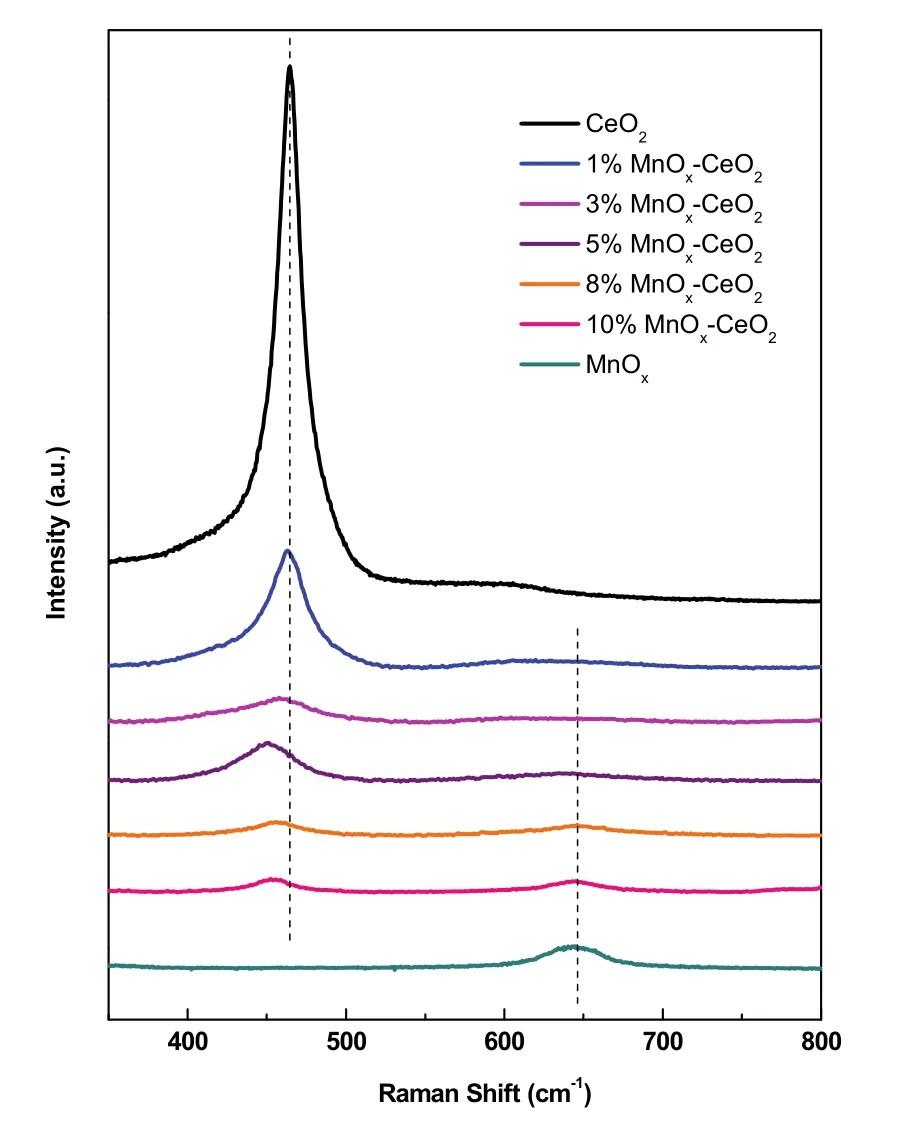
图8 w MnO x-CeO2的可见拉曼图Fig. 8 Visible Raman of w MnO x-CeO2.
图9 是w MnOx-CeO2催化剂的紫外激发Raman谱图。由于紫外光激发材料表面产生拉曼散射光时会发生紫外共振效应加强拉曼信号,因此紫外拉曼对缺陷有更高的灵敏性。图9中除了观察到465 cm−1处的F2g特征峰,还出现了位于600 cm−1的归属于CeO2填隙式氧空位22的特征峰,以及位于1179 cm−1归属于二阶纵向声子峰的特征峰。根据文献报道25,归属于填隙式氧空位的600 cm−1的特征峰峰面积与F2g峰465 cm−1的特征峰面积的比值,可反映晶体表面氧空位的浓度。图10是w MnOx-CeO2的A600/A465值即表面氧空位浓度图,可以看出,MnOx-CeO2相对于单一的CeO2具有更高浓度的氧空位,且浓度随着Mn含量的增加呈现上升趋势,直至达到5%后氧空位浓度基本保持不变,这与可见拉曼的结果一致。这说明Mn的引入,提高了催化剂表面氧空位浓度。同时不难看出,催化剂的活性与表面氧空位浓度正相关。

图9 w MnO x-CeO2的紫外拉曼图Fig. 9 UV Raman of w MnO x-CeO2.
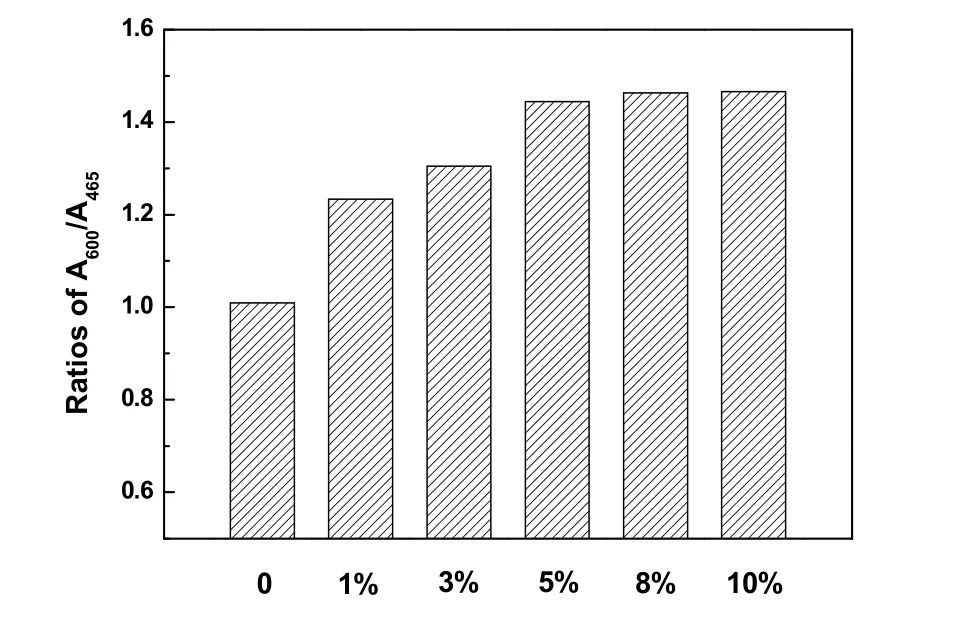
图10 w MnO x-CeO2的表面氧空位浓度图Fig. 10 Oxygen vacancies concentration of w MnO x-CeO2.
3.3.4 XPS
至此,XRD、BET、SEM-EDS、HRTEM和Raman等表征都证实了:引入Mn的过程中,除了部分Mn进入CeO2晶格形成固溶体外,其余Mn主要分散在CeO2表面,且分为单层分散态和晶相态。当Mn载量超过单层分散阈值时,催化剂体系物相结构发生改变,体系的许多性质也会在阈值处出现转折,即所谓的“阈值效应”,其直接影响催化剂活性。因此对本体系中单层分散阈值这一关键参数的研究至关重要。
XPS峰强度法,是通过测定准表面内元素含量来推求单层分散阈值的有效方法24。为了更准确地估算阈值,本文对Mn含量从1%到30%样品进行了XPS表征,得到了Mn 2p和Ce 3d的峰面积IMn2p和ICe3d,并且把1%–30%的Mn质量分数转化为对应的Mn载量(gMn3O4/gCeO2,即单层分散阈值的常用单位),随后对数据进行分析。Mn载量与峰比值的递变关系如图11所示。图中直线1斜率大,在这个线段范围内,Mn3O4以单层形式高度分散在CeO2表面上,Mn2+/Mn3+和O2−通过离子键力和CeO2表面的O2−和Ce4+相互作用形成表面化合物26。而直线2斜率突然变小,说明此时单层分散后剩余的Mn3O4是以晶粒形式存在的,其均匀地分布在已铺设Mn3O4单层的CeO2载体表面上。两条直线的交点所对应的Mn载量是0.75 gMn3O4/gCeO2,换算为Mn质量分数是6.2%。此交点被认为是Mn3O4在载体表面呈单层分散和出现晶相的分界线27,也就是单层分散阈值。
在Mn质量分数达到6.2%之后,催化剂体系物理化学性质发生变化,与样品在XRD,BET,HRTEM和Raman等表征中表现出的结构性质转折点相一致。此时催化剂活性不再随着Mn的增加而提高,基本保持不变24。
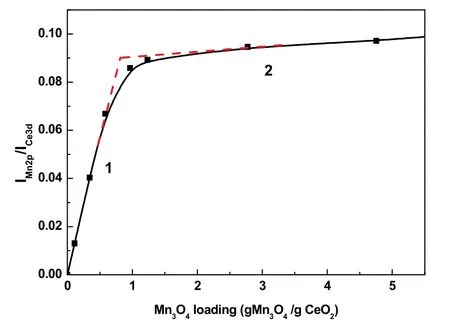
图11 I Mn2p/I Ce3d比值和Mn载量的关系Fig. 11 Relationship between ratios of I Mn 2p/I Ce 3d and Mn loading.
为了进一步研究催化剂表面各元素价态以及反应过程中的氧物种,对Mn含量最接近单层分散阈值的5%MnOx-CeO2催化剂反应前后的样品进行XPS分析,结果见图12和表3。在Ce 3d谱图中,以u和v标记的特征峰属于Ce的3d3/2和3d5/2轨道,其中归属于Ce4+的特征峰包括v,v2,v3,u,u2和u3,归属于Ce3+的特征峰为v1和u128,29。反应前后Ce4+/Ce3+的比值为4.97和4.47(> 1),说明Ce主要以+4价态存在。在Mn 2p谱图中,641.2 eV和651.6 eV的特征峰归属于Mn2+和Mn3+,而642.6 eV和653.1 eV附近的特征峰则归属于Mn4+30。可见,催化剂表面主要以Mn2+和Mn3+为主。谱图中相应峰的结合能(641.6 eV和653.0 eV)都比文献高,表明MnOx在CeO2上的高度分散状态31,同时也说明其与载体之间存在相互作用从而引起化学位移32,33。在O 1s谱图中,结合能在533–534 eV间的特征峰归属为吸附氧(Oads),在530–532 eV间为表面氧(Osur),在529–530 eV间是晶格氧(Olatt)30,34。从表中可以看出,催化剂反应之后Mn4+/(Mn2++ Mn3+)和Ce4+/Ce3+的比值都出现了下降的现象,说明这些物种都参与到了催化体系的氧化还原循环当中。而氧物种中,Olatt反应后数目减少,Osur和Oads则数目增多,推断是由于反应过程中Olatt和Osur的不断消耗,致使气相中的O2被吸附到催化剂表面氧空位处补充活性氧物种35,从而形成促进反应的氧循环。综上所述,在催化甲苯的反应中,Mn4+/(Mn2++Mn3+),Ce4+/Ce3+和Olatt,Osur都参与到氧化还原过程当中,这一结论有助于更微观地阐明催化反应过程。
3.3.5 H2-TPR表征

图12 反应前后5%MnO x-CeO2的XPS谱图Fig. 12 XPS spectra of fresh and reacted 5%MnO x-CeO2.

表3 5%MnO x-CeO2的XPS表面元素分析Table 3 Atomic ratios by XPS surface compositional analysis for 5%MnO x-CeO2.
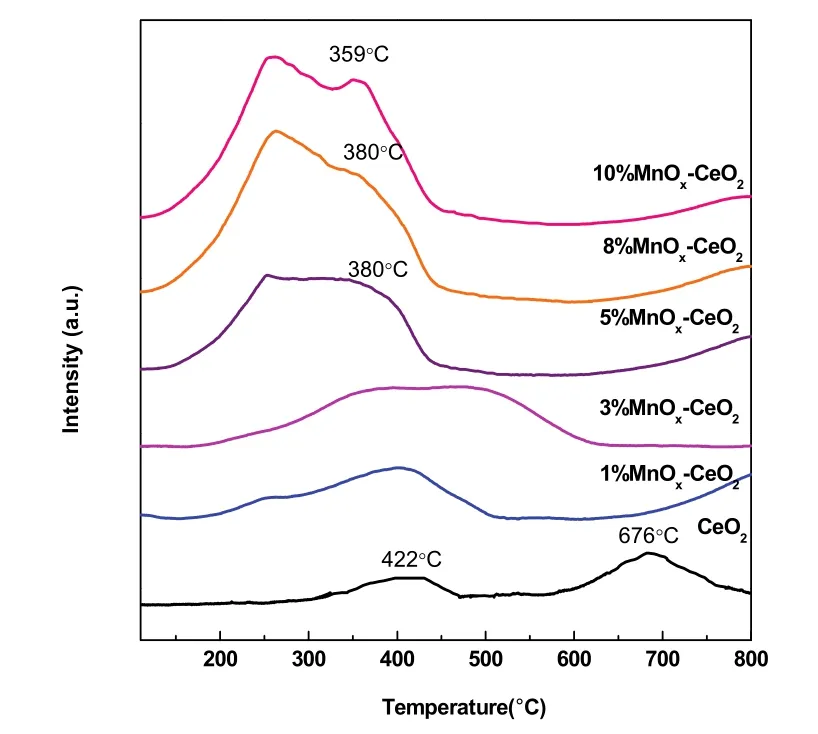
图13 w MnO x-CeO2的H 2-TPR图Fig. 13 H 2-TPR of w MnO x-CeO2.

表4 w MnO x-CeO2的还原峰位置及耗氢量Table 4 Position of reduction peaks and H2 consumption for w MnO x-CeO2.
w MnOx-CeO2催化剂的氧化还原性能利用H2-TPR来表征,各样品的还原峰如图13和表4所示。观察CeO2的还原峰,在422 °C的低温峰可归属于体表CeO2向Ce2O3的还原,而676 °C的特征峰则归属于体相CeO的还原36。对于Mn含量低的MnOx-CeO2,其特征峰主要位于250–600 °C温度区间内,呈现一个大范围的宽包,此还原峰归属为单层分散态的锰由高价态向低价态的转换37–39。当Mn含量增加时,即单层分散态Mn含量不断增加,此还原峰的峰面积随分散量的增加逐渐增加。当Mn含量进一步增大直至接近或超过单层分散阈值6.2%这一临界点时,还原峰发生低温偏移,同时在较高温度(约380 °C)出现一个新的还原峰19。结合XRD,Raman和XPS等表征结果以及文献报道40,这个新的还原峰归属为晶相态Mn3O4物种的还原。这一表征结果与催化剂活性规律保持一致,能很好地解释分析影响活性的关键组分:在Mn含量低的情况下,Mn以单层分散形式存在,表面化合物的氧化还原能力较弱,此时活性较低;随着Mn含量的提高,表面化合物含量也增加,其氧化还原能力逐渐增强,此时活性得到提高;当达到单层分散阈值的时候,此时表面化合物数目不再增加,催化剂的氧化还原性能达到最大值,活性也最高;而当超过单层分散阈值时,Mn晶相态出现,而氧化还原性能和活性也基本保持不变。综上所述,锰物种和CeO2的相互作用会削弱Ce―O键从而增强其表面氧的还原性;其中,分散态Mn和晶相态Mn的还原性能表现出明显差异,前者比后者在热力学上更稳定;在本体系中,决定着催化剂的活性大小的重要因素是Mn的单层分散化合物。
3.3.6 UV-Vis表征
为了进一步探讨单层分散化合物在载体表面的分布状态,以及此活性组元影响催化活性的本质机理,对催化剂进行UV-Vis表征,以获得Mn物种在 CeO2表面分布状态的信息,结果如图 14所示。可以观察到在250 nm处有一个明显的峰,主要归因于载体中 O2−到表面 Mn3+的电子转移跃迁,而550 nm处较为微弱的峰,则归属于Mn2+晶体场的6A1g→4T2g跃迁41。因此可得出以下结论,一是 Mn物种和载体之间存在强相互作用而发生电子迁移和交换,二是表面 Mn主要以 Mn2+和Mn3+形式存在。根据文献报道42,二者的强相互作用可用嵌入模型来解释。该模型认为在适当条件下,Mn活性组元在CeO2表面分散作用的实质是 MnOx的金属阳离子进入载体表面的晶格立方空位,而与之相伴的氧阴离子则处于这些阳离子占据的位置上,以抵消过剩的正电荷。通过TEM表征可知CeO2优先暴露(111)晶面,因此MnOx在CeO2表面的分散可用图15示意。由此可知,Mn活性组分影响活性的本质机理是:Mn以间隙离子形式填入晶格空位,引起晶格畸变,诱导形成氧空位,表面氧活动性增加,因此造成催化剂氧空位浓度增大,从而有效促进催化剂的还原,提高活性43。这与上文的XRD,Raman和H2-TPR结果相一致;另外随着Mn含量增加至单层分散阈值,CeO2表面已没有多余的晶格空位容纳 Mn离子,因此活性不再得到提高。
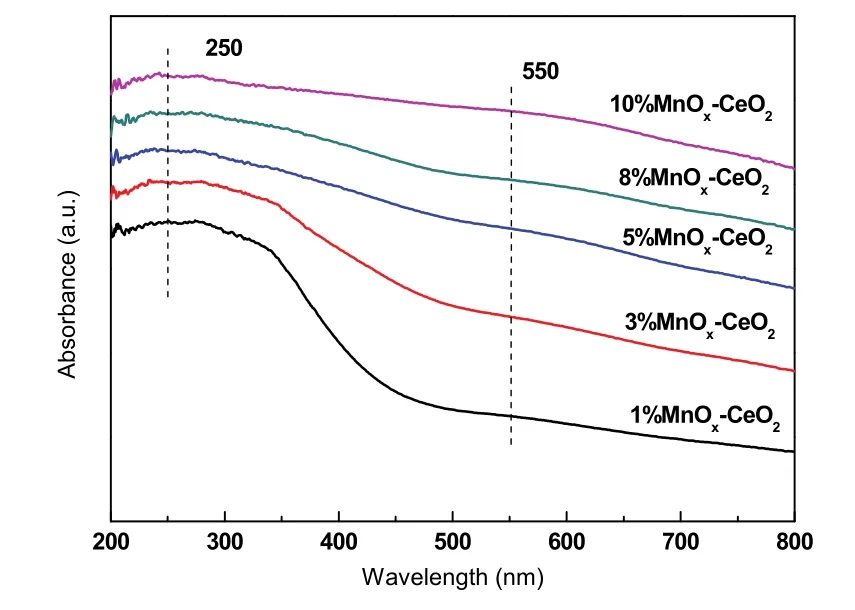
图14 w MnO x-CeO2的 UV-Vis图Fig. 14 UV-Vis of w MnO x-CeO2.
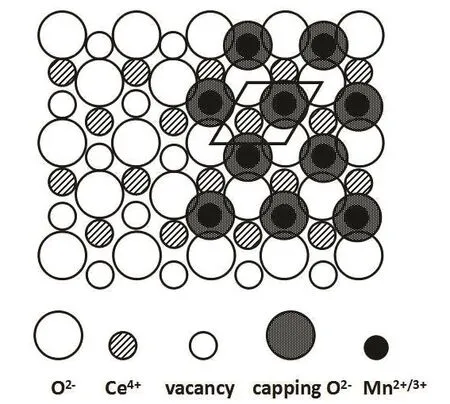
图15 MnO x在CeO2(111)面上分散的示意图Fig. 15 Diagram of MnO x supported on the surface of CeO2(111).
4 结论
采用MOF模板法制备了系列不同Mn含量的MnOx-CeO2催化剂,且具备棒状形貌、高度分散、高比表面积和纳米晶体颗粒等特征。在引入Mn的过程中,除了部分Mn进入CeO2晶格形成固溶体外,其余Mn主要分散在CeO2表面,且分散的Mn分为单层分散态和晶相态。当Mn载量低于或等于单层分散阈值6.2%时,Mn主要以嵌入模型的形式与表面CeO2发生强相互作用,引起电荷转移和交换,诱导氧空位形成使催化剂氧空位浓度增大,从而有效促进催化剂的还原,提高活性;当Mn载量大于单层分散阈值6.2%时,在表面单层分散化合物上Mn3O4微晶逐渐生成,对活性无明显促进作用。这些结果表明,在催化剂制备中,将活性组份氧化物的含量控制在接近单层分散阈值,有利于提高催化剂的活性。
(1) Kim, K.; Ahn, H. Appl. Catal. B: Environ. 2009, 91, 308.doi: 10.1016/j.apcatb.2009.05.037
(2) Li, P.; He, C.; Cheng, J.; Ma, C.; Dou, B.; Hao, Z. Appl. Catal. B:Environ. 2011, 101, 570. doi: 10.1016/j.apcatb.2010.10.030
(3) Liotta, L. Appl. Catal. B: Environ. 2010, 100, 403.doi: 10.1016/j.apcatb.2010.08.023
(4) De Rivas, B.; Fonseca, R.; Sampedro, C.; Gutiérrez-Ortiz, J. Appl.Catal. B: Environ. 2009, 90, 545. doi: 10.1016/j.apcatb.2009.04.017
(5) Okal, J.; Zawadzki, M. Appl. Catal. B: Environ. 2009, 89, 22.doi: 10.1016/j.apcatb.2008.11.024
(6) Wu, X.; Liu, S.; Weng, D.; Lin, F.; Ran, R. Hazard. Mater. 2011,187, 283. doi: 10.1016/j.jhazmat.2011.01.010
(7) Tikhomirov, K.; Krocher, O.; Elsener, M.; Wokaun, A. Appl. Catal.B: Environ. 2006, 64, 72. doi: 10.1016/j.apcatb.2005.11.003
(8) Becerra, M.; Arias, N.; Giraldo, O.; Suarez, F.; Gomez, M.; Lopez,A. Appl. Catal. B: Environ. 2011, 102, 260.doi: 10.1016/j.apcatb.2010.12.006
(9) Santos, V.; Pereira, M.; Orfao, J.; Figueiredo, J. Appl. Catal. B:Environ. 2010, 99, 353. doi: 10.1016/j.apcatb.2010.07.007
(10) Bastos, S.; Orfao, J.; Freitas, M.; Pereira, M.; Figueiredo, J. Appl.Catal. B: Environ. 2009, 93, 30. doi: 10.1016/j.apcatb.2009.09.009
(11) Shan, W.; Ma, N.; Yang, J.; Dong, X.; Liu, C.; Wei, L. J. Nat. Gas Chem. 2010, 19, 86. doi: 10.1016/S1003-9953(09)60033-5
(12) Champness, N.; Schroder, M. Curr. Opin. Solid. St. M. 1998, 3, 419.doi: 10.1016/S1359-0286(98)80055-7
(13) Zhang, L.; Wu, H.; Madhavi, S.; Hng, H.; Lou, X. J. Am. Chem. Soc.2012, 134, 17388. doi: 10.1021/ja307475c
(14) Fuertes, A.; Centeno, T. J. Mater. Chem. 2005, 15, 1079.doi: 10.1039/b416007j
(15) Das, R.; Pachfule, P.; Banerjee, R.; Poddar, P. Nanoscale 2012, 4,591. doi: 10.1039/c1nr10944h
(16) Zhang, F.; Chen, C.; Xiao, W.; Xu, L.; Zhang, N. Catal. Commun.2012, 26, 25. doi: 10.1016/j.catcom.2012.04.028
(17) Xu, L.; Chen, C.; Wang, R.; Luo, J.; Liu, Y.; Zhang, N. Chem. J.Chin. Univ. 2013, 34, 1907. [徐力, 陈超, 王瑞, 罗家还, 刘云凌,张宁. 高等学校化学学报, 2013, 34, 1907.]doi: 10.7503 /cjcu20130260
(18) Liu, K.; You, H.; Jia, G.; Zheng, Y.; Huang, Y.; Song, Y.; Yang, M.;Zhang, L.; Zhang, H. Cryst. Growth Des. 2010, 10, 790.doi: 10.1021/cg901170j
(19) Dai, Y.; Li, S.; Tang, C.; Yao, X.; Qi, L.; Liu, B.; Gao, F.; Dong, L.Chin. J. Inorg. Chem. 2012, 28, 1555. [戴越, 李珊珊, 汤常金, 姚小江, 齐蕾, 刘斌, 高飞, 董林. 无机化学学报, 2012, 28, 1555.]
(20) Wang, C.; Zhao, B.; Xie, Y. Chin. J. Catal. 2003, 24, 475. [王春明,赵壁英, 谢有畅. 催化学报, 2003, 24, 475.]
(21) Kaliaguine, S.; Van Neste, A.; Szabo, V.; Gallot, J.; Bassir, M.;Muzychuk, R. Appl. Catal. A: Gen. 2001, 209, 345.doi: 10.1016/S0926-860X(00)00779-1
(22) Wu, Z.; Li, M.; Howe, J.; Meyer, H.; Overbury, S. Langmuir 2010,26, 16595. doi: 10.1021/la101723w
(23) Hungria, A.; Fernandez-Garcia, M.; Anderson, J.; Martinez-Arias, A.J. Catal. 2005, 235, 262. doi: 10.1016/j.jcat.2005.08.012
(24) Deng, C.; Duan, L.; Xu, X.; Xie, Y. Nat. Gas Chem. Ind. 1992, 6, 6.[邓存, 段连运, 徐献平, 谢有畅. 天然气化工, 1992, 6, 6.]
(25) Taniguchi, T.; Watanabe, T.; Sugiyama, N.; Subramani, A.; Wagata,H.; Matsushita, N.; Yoshimura, M. J. Phys. Chem. C 2009, 46,19789. doi: 10.1021/jp9049457
(26) Liu, Y.; Wu, J.; Guo, Q.; Gui, L.; Tang, Y. Chin. J. Catal. 1987, 8,14. [刘英骏, 吴江平, 郭沁林, 桂琳琳, 唐有祺. 催化学报, 1987,8, 14.]
(27) Gui, L.; Liu, Y.; Guo, Q.; Huang, H.; Tang, Y. Sci. China Chem.1985, 6, 509. [桂琳琳, 刘英骏, 郭沁林, 黄惠忠, 唐有祺. 中国科学, 1985, 6, 509.]
(28) Zhang, Y.; Zhang, L.; Deng, J.; Dai, H.; He, H. Inorg. Chem. 2009,48, 2181. doi: 10.1021/ic802195j
(29) Li, Y.; Sun, Q.; Kong, M.; Shi, W.; Huang, J.; Tang, J.; Zhao, X.J. Phys. Chem. C 2011, 115, 14050. doi: 10.1021/jp202720g
(30) Kan, J.; Deng, L.; Li, B.; Huang, Q.; Zhu, S.; Shen, S.; Chen, Y.Appl. Catal. A: Gen. 2017, 530, 21.doi: 10.1016/j.apcata.2016.11.013
(31) Wan, H.; Li, D.; Dai, Y.; Hu, Y.; Liu, B.; Dong, L. J. Mol. Catal.A -Chem. 2010, 332, 32. doi: 10.1016/j.molcata.2010.08.016
(32) Di Monte, R.; KasPar, J.; Fornasiero, P.; Graziani, M.; Paze, C.;Gubitosa, G. Inorg. Chim. Acta 2002, 334, 318.doi: 10.1016/S0020-1693(02)00800-9
(33) Qi, G.; Yang, R. J. Phys. Chem. B 2004, 108, 40.doi: 10.1021/jp048431h
(34) Wang, X.; Kang, Q.; Li, D. Appl. Catal. B: Environ. 2009, 86, 166.doi: 10.1016/j.apcatb.2008.08.009
(35) Rico-Pérez, V.; Aneggi, E.; Bueno-López, A.; Trovarelli, A. Appl.Catal. B: Environ. 2016, 197, 95. doi: 10.1016/j.apcatb.2016.02.051(36) Liao, Y.; Fu, M.; Chen, L.; Wu, J.; Huang, B.; Ye, D. Catal. Today 2013, 216, 220. doi: 10.1016/j.cattod.2013.06.017
(37) Lin, X.; Li, S.; He, H.; Wu, Z.; Wu, J.; Chen, L.; Ye, D.; Fu, M.Appl. Catal. B: Environ. 2017, doi: 10.1016/j.apcatb.2017.06.071
(38) Liu, X.; Li, S.; Sun, M.; Yu, C.; Huang, B. Acta Phys. -Chim. Sin.2016, 32, 1236. [刘小青, 李时卉, 孙梦婷, 喻成龙, 黄碧纯. 物理化学学报, 2016, 32, 1236.] doi: 10.3866/PKU.WHXB201602251
(39) Li, D.; Yu, Q.; Li, S.; Wan, H.; Liu, L.; Qi, L.; Liu, B.; Gao, F.;Dong, L.; Chen, Y. Chem.-A Eur. J. 2011, 17, 5668.doi: 10.1002/chem.201002786
(40) Zhang, Y.; Xie, Y.; Zhang, Y.; Zhang, D.; Tang, Y. Sci. China Chem. 1986, 8, 805. [张玉芬, 谢有畅, 张阳, 张德龙, 唐有祺.中国科学, 1986, 8, 805.]
(41) Parida, K.; Dash, S.; Singha, S. Appl. Catal. A: Gen. 2008, 351, 59.doi: 10.1016/j.apcata.2008.08.027
(42) Yu, Q.; Zhang, S.; Wang, X. Ind. Catal. 2007, 15, 12. [郁青春, 张世超, 王新东. 工业催化, 2007, 15, 12.]
(43) Dong, L.; Chen, Y. Chinese J. Inorg. Chem. 2000, 2, 250. [董林,陈懿. 无机化学学报, 2000, 2, 250.]
