聚酚氧/环氧树脂共改性氰酸酯树脂的制备及其低温性能
2018-06-09张海琪高堂铃吴健伟付春明
张海琪, 王 冠,2, 高堂铃,2, 付 刚,2, 吴健伟,2,匡 弘,2, 付春明,2
未来新一代先进航天飞行器运行环境更加苛刻(–200~200 ℃),这就要求某些特殊结构的复合材料需在较宽的温差范围内具有尺寸和强度的稳定性,而现今航天工程中大量应用的环氧树脂基复合材料,由于其树脂基体的耐温性较差(Tg<200 ℃),已不适合于先进航天器某些特殊结构的复合材料设计[1-3]。近年来,氰酸酯树脂(CE)由于固化后形成独特的三嗪环结构并且交联点间通过醚键相连,具有优异的耐高温稳定性(Tg>300 ℃)、良好的高、低温力学强度和工艺性能,具备了可在深空环境稳定应用的潜质[4-5]。由于氰酸酯树脂交联密度较大,单独使用时其固化物的韧性仍然较差,从而影响了在低温环境中的应用效果,因此,需要对氰酸酯树脂进行改性提高其低温韧性。
现阶段增韧改性氰酸酯的方法通常是在氰酸酯网络中引入液体橡胶或热塑性工程塑料,利用固化微分相技术可大幅度地提高氰酸酯树脂的韧性,其中室温韧性可提高2.5~3.2倍[6];但是,通过上述方法改性得到的氰酸酯固化物经室温至超低温(–150 ℃以下)冷热循环过程时, 在两相界面产生内应力,过多内应力积累超过树脂本身的强度导致树脂基体的破坏,从而达不到低温增韧效果[7]。目前,国内外对于氰酸酯树脂低温增韧改性方法和机理的研究尚属于起步阶段,相关文献鲜有报道。本研究团队王冠等[8]采用了高玻璃化温度的特种聚醚砜树脂(PES-C)和高纯环氧树脂共改性氰酸酯树脂,得到的固化物在–100~200 ℃范围内获得了较高的韧性和抗拉强度,但是没有提及在–200 ℃超低温时改性氰酸酯树脂的韧性和力学强度数据。
聚酚氧树脂(PKHH)是一种超高分子量(5万~40万)的环氧树脂,主链结构中含有大量的醚键,侧链含有羟基重复单元,赋予了树脂较好的机械强度和低温韧性,与极性树脂基体有较好的相容性。有文献报道[9-10],采用PKHH改性环氧树脂获得了很好的低温(–150 ℃)增韧效果,但是相关的数据和增韧机理并未提及。本研究采用PKHH树脂和低黏度高纯环氧树脂(EP)共改性氰酸酯树脂,研究其混合物的固化行为。考察改性后的氰酸酯树脂在–196 ℃环境下力学性能和界面断裂形貌,分析其低温增韧机理和低温力学强度的影响因素,考察改性后氰酸酯树脂的耐热性和经高低温冷热循环(–196 ~200 ℃)后的耐久性等性能。
1 实验
1.1 原料
双酚A型氰酸酯(CE),工业级,吴桥树脂厂;环氧树脂 (EP),DER354,环氧当量(g/eq)168~182,陶氏化学公司;聚酚氧(PKHH),= 52000 g/mol,美国联碳公司。
1.2 试样配比及制备
PKHH改性CE/EP混合物各组分配比见表1,其中不同试样的PKHH含量是可变的,CE/EP的质量比维持约为4.7:1不变。按照表1中配比要求(除CYEP0试样外),将PKHH加入到130 ℃的EP中,待完全溶解后将其加入已溶解的CE中,升温至(150 ± 5)℃,高速搅拌(1000~1500 r/min),维持2~3 h,得到均一透明的液体,将其倒入已预热好(150 ℃)的模具中,真空脱泡,按照150 ℃/2 h +180 ℃/4 h + 200 ℃/4 h + 230 ℃/2 h 程序化升温固化后,得到四种不同配比的固化物分别为CEPK0,CEPK5,CEPK10,CEPK15。
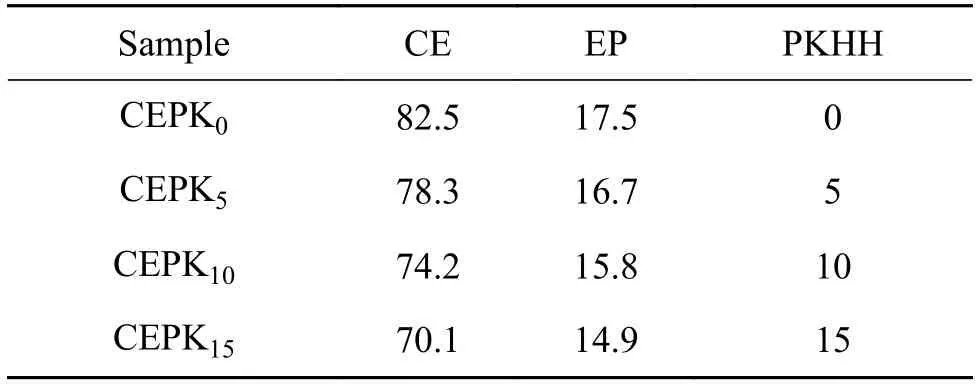
表1 PKHH改性CE/EP混合物配比表(质量分数/%)Table 1 Blend ratio of CE/EP modified by PKHH(mass fraction/%)
1.3 分析与测试
差示扫描量热分析(DSC):采用Q20型差示扫描量热分析仪测定反应热焓和固化曲线,升温速率10 ℃/min,扫描温度为40~350 ℃。固化度α计算方法见式(1)[8,11]:
式中:α 为固化度;ΔH 为反应总热焓,J·g–1;ΔHr为反应剩余热焓,J·g–1。
红外光谱分析(FTIR):采用VECTOR-22型傅立叶红外光谱仪,KBr压片法,扫描范围为4000~500 cm–1。
力学性能测试:采用INSTRON-4467型万能材料试验机和摆锤冲击试验机,按GB/T 2567—2008相关测试方法对试样的拉伸性能、弯曲性能和冲击韧性进行测试。热老化测试是将拉伸试样放置在150 ℃老化箱中,经过100 h老化后, 取出放置到室温,测定拉伸强度。
动态热机械分析:采用DMS 6100动态机械热分析仪,测试模式双悬臂,试样尺寸为50 mm ×10 mm × 3 mm,振动频率为1 Hz,扫描范围为–150~250 ℃,升温速率为 5 ℃/min。
扫描电镜测试:对待测样品断面进行喷金处理,采用SU3500型扫描电子显微镜观察试样断面形貌。
热失重分析:采用6300型热失重分析仪测定试样的热分解特性,测试样品质量约5 mg,空气氛围,升温速率为 10 ℃/min,温度范围 50~700 ℃。
低温性能测试:测定树脂浇铸体试样在–196 ℃下的拉伸强度、弯曲强度和冲击韧性。拉伸强度、弯曲强度和冲击韧性的测试方法和试样尺寸均参照GB/T 2567—2008的相关规定执行,将处理好的拉伸和弯曲试样放置在自制低温保温套件中,保温10 min后开始测试。冲击韧性测试:试样在装有液氮的自制保温套件中保温10 min后,取出5 s内完成测试。
冷热循环测试:试样尺寸为35 mm × 5 mm。实验前将试样放置于50 ℃干燥箱中,恒温30 min,然后取出,迅速将其放入装有液氮的保温器皿中,保温10 min,取出放入200 ℃恒温箱内保温10 min,冷-热循环后测定试样表面的裂纹密度(根/cm2)。
2 结果与讨论
2.1 PKHH改性CE/EP混合物的固化行为
利用DSC和FTIR考察PKHH改性CE/EP混合物(CEPK树脂)的固化行为。图1(a),(b)为混合物固化前后的DSC曲线。由图1可知,随着CE/EP混合物中PKHH含量的增加,混合物的反应放热峰向低温区移动,DSC曲线形状不变,混合物固化后的反应放热焓逐渐减少,含有15%PKHH的CE/EP混合物(CEPK15)相对于未添加PKHH的混合物(CEPK0)的反应起始温度降低约40 ℃,说明PKHH的加入可明显促进CE/EP混合物的固化反应。
表2为不同PKHH含量改性CE/EP混合物固化反应参数。由表2进一步证明,经规定的固化工艺固化后,含有PKHH的试样的固化度α明显提高,并且随着PKHH的增加固化度进一步提高,其中CEPK15固化度接近100%。
图2为CEPK0和CEPK10试样经过180 ℃等温固化过程的FTIR谱图。在图2(a),(b)中观察到,经过180 ℃/3 h后,CEPK10中的氰酸酯基特征峰(2270~2235 cm–1)消失,环氧基特征峰(915 cm–1)减弱,同时出现噁唑烷酮特征峰(1762 cm–1),而不含PKHH的混合物CEPK0经过180 ℃/3 h后仍有氰酸酯单体和环氧基团存在,仅有噁唑啉结构出现并且未见噁唑烷酮吸收峰,经过6 h固化后出现噁唑烷酮结构。这些结果进一步证实,PKHH的存在促进了氰酸酯单体的自聚和与环氧树脂插入共聚成环的反应。另外,对比CEPK0和CEPK10的FTIR谱图,两者的特征峰的位置相似,且得到的最终固化物红外图谱基本保持一致,可初步推断PKHH的存在并未改变CE/EP最终产物结构。
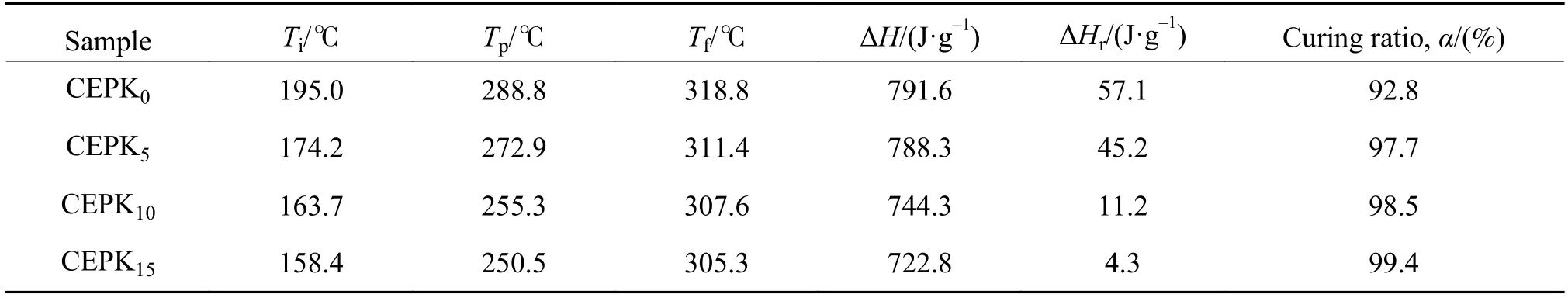
表2 不同PKHH含量改性CE/EP混合物固化反应参数表Table 2 Curing reaction data of CE/EP blends with different PKHH contents
2.2 PKHH改性CE/EP混合物的增韧机理
利用SEM观察PKHH改性CE/EP固化物后的断面微观形貌,结果如图3。图3(a),(b)为CEPK0和CEPK15在24 ℃下冲击断面微观形貌图,(c),(d)为–196 ℃下冲击断面微观形貌图。由图3(a),(b)看出,24 ℃下随着PKHH的增加,CE/EP断面粗糙度增加,出现河流状纹路且韧窝增多,裂纹扩展被束缚并出现弯曲和分叉,呈现韧性断裂。这表明PKHH的加入能明显提高CE/EP混合物抵抗外载荷的能力。进一步放大(a),(b)图观察发现,PKHH改性CE/EP混合物固化后,并没有出现相分离形成球状分散相或发生相反转,这说明线性的PKHH长链分子在CE/EP固化网络中有较好的相容性,与通用热塑性树脂(如聚砜、聚苯乙烯等)改性CE树脂通过分散相粒子吸收冲击能量提高韧性的增韧机理不同[12]。因此,推断PKHH与CE/EP混合物形成半互穿网络结构(SIPN)[13-15],可能的增韧机理为PKHH的多羟基结构与CE树脂极性相近,通过PKHH长链分子链缠绕的“协同效应”和“强迫包容”作用提高了CE/EP混合物的韧性[16]。由图3(c),(d)看出,在–196 ℃条件下,未加入PKHH的CEPK0断面出现了贯通的河流状裂纹,而在室温下其断面较光滑,这可能是由于超低温下分子链段振动大为减缓,分子间的堆积更加紧密,分子间作用力加大,因此,表现出了一定的韧性断裂特征。与室温下增韧规律相似,在超低温下(–196 ℃),由于PKHH的多醚键结构比CE/EP活动能力更大,进一步增加了CE/EP混合物抵消冲击载荷的能力,因而CEPK15体现出了更好的韧性。
DMA测试方法是通过对高分子材料施加一定频率的正弦载荷获取材料的动态力学响应,包括储能模量、损耗模量和阻尼系数,它们的峰值变化对应材料的内部结构变化,相应的温度为转变特征温度。因此,通过DMA法进一步研究了PKHH改性CE/EP固化物的网络构成和韧性关系。表3为PKHH改性CE/EP混合物不同温度下的冲击韧性。图4为不同PKHH含量的CE/EP混合物的损耗模量曲线。由图4可知,α峰为混合物固化网络中分子链段松弛转变峰,β峰为分子基团、链节自旋松弛次级转变峰[17]。CE/EP混合物中随着PKHH的增多,α峰和β峰均有向低温移动的趋势且β峰的峰宽变大,除CEPK0外其他试样均发现一个α峰。上述现象可能的解释如下:(1)PKHH存在与CE/EP树脂的插入反应,对CE固化网络具有一定的溶胀作用,导致交联点间的距离增加和耐热性降低,因此体现出α峰向低温转移;(2)由于PKHH的多醚键结构(具有低温活动能力),使得在超低温下CE树脂固化网络中可活动的基团、链节数增多,导致了β峰向更低温度区域移动;(3)尽管PKHH的玻璃化温度与CE树脂的相差较大,由于PKHH自身多羟基的特点,CE树脂固化网络对其产生了“强迫包容”作用,增加了两者间的结合力,表现出了仅有一个α峰。综合表3和图4的分析结果,固化物的韧性提高与在一定温度下固化网络分子链段的消耗能量的能力有关,因此,PKHH的加入从微观结构上提升了CE/EP固化物的韧性,其中CEPK15表现出较好的高低温韧性。另外还发现,CEPK0的α峰相邻处有一伴峰存在,结合表2数据可知,CEPK0的固化度仅有92.8%,含有较多的未完全固化组分,推测这些未完全固化的组分可能是氰酸酯形成的低聚体或残余单体,因此,在DMA曲线上明显地表现出与主网络结构不同的热-机械特性,即在α峰附近出现伴峰。
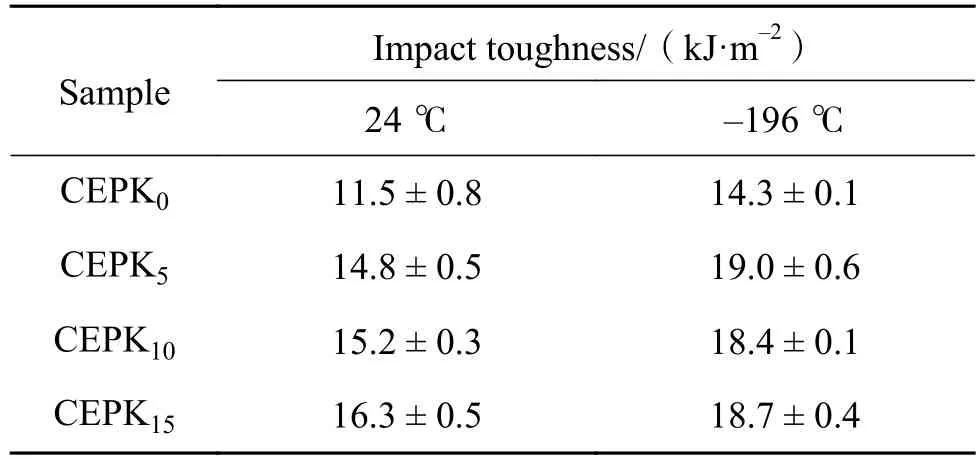
表3 PKHH改性CE/EP混合物冲击韧性Table 3 Impact toughness of PKHH modified CE/EP blends
2.3 PKHH改性CE/EP固化物力学性能
图5(a), (b)分别为PKHH改性CE/EP固化物在室温和–196 ℃下的力学性能变化曲线。由图5(a), (b)看出,室温条件下,随着PKHH含量的增加,CE/EP混合物的力学强度呈现先增高后略微下降的趋势,而固化物的模量几乎不受PKHH含量的影响,维持在3.5 GPa左右,说明适量的PKHH能有效改善CE固化物的缺陷,提高力学强度。拉伸强度最大提高20%,而不会改变CE树脂的固化网络基本结构,也进一步证实了PKHH通过“SIPN”的方式对CE固化物进行改性。在超低温条件下,随着PKHH的增加,CE/EP固化物的拉伸强度变化与室温下测得的结果略有不同,其逐步增加并不存在下降趋势,而模量变化规律不明显但总体数值高于室温测试值约30%,这可能是由于PKHH的醚键低温活动能力强和PKHH未发生相分离增韧有关。综合考虑,10%~15%的PKHH改性CE/EP混合物能有效改善氰酸酯树脂的低温力学性能而不影响CE树脂的刚性。
2.4 PKHH改性CE/EP固化物耐热性能
从固化物网络结构热稳定性角度出发,利用TGA考察了PKHH改性CE/EP固化物的热稳定特性。图6为不同PKHH含量改性CE/EP混合物的TGA曲线。由图6可知,几种混合物的热失重曲线形状相似,与文献报道的纯CE树脂的热失重历程相同[18],即主要分三阶段:第一拐点是5%失重左右,主要是碳氢断链,解交联等,此时温度为初始热分解温度(IDT);第二拐点是10%~50%失重,主要降解反应,三嗪环降解;最后是60%~100%失重,主要是残碳与氧,氮等元素反应气化,这说明PKHH和EP的加入不会改变CE固化网络的主体结构。随着PKHH含量的增多,CE/EP混合物的IDT略下降,但是不会对CE/EP固化网络的热稳定性造成严重影响。
通过对 CEPK0,CEPK5,CEPK10,CEPK15试样的高温力学强度和高温老化性能测试,考察PKHH改性CE/EP混合物在实际应用环境中的热稳定性和耐久性。表4为PKHH改性CE/EP固化物不同温度下的拉伸性能,图7为PKHH改性CE/EP固化物经热老化后的拉伸性能。由表4 可知,随着PKHH增加CE/EP固化物在150 ℃的强度基本相同,强度保持率均大于85%,同时发现200 ℃强度下降较多,强度保持率仅有30%~40%,且数值变化无规律,这主要是因为200 ℃测试条件接近混合物的玻璃化温度,导致网络分子活动能力增加,抵抗外载荷的能力下降。由图7可知经过150 ℃热老化100 h后,PKHH改性的CE/EP固化物经热老化后强度保持率达到90%以上。
综合分析,在玻璃化转变温度以下50 ℃附近,PKHH改性CE/EP固化物具有较好的热稳定性和耐久性,含有5%~10%的PKHH的固化物在200 ℃具有较高的强度。
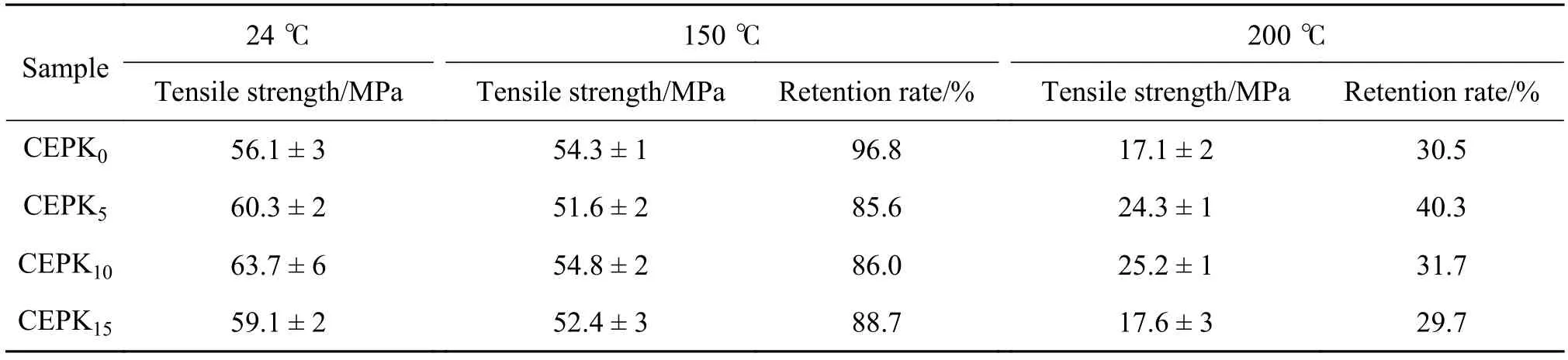
表4 PKHH改性CE/EP混合物不同温度下的拉伸性能Table 4 Tensile properties of PKHH modified CE/EP blends at different temperatures
2.5 PKHH改性CE/EP固化物超低温耐久性能
在深空低温环境中,材料实质上需要经过低温-高温的苛刻历程,因此,考察材料耐冷-热交变特性对于研究材料在超低温环境中耐久应用的适用性是必要的。图8为CEPK10试样经冷热循环30次后的形貌。表5为不同树脂固化物经冷、热循环后材料首次出现裂纹情况。由表5可知,在CE树脂中随着PKHH的增多,混合物的耐冷-热交变性能提高,但是过多的PKHH加入后,CE树脂的耐低温稳定性反而略有降低,主要是因为超过15%PKHH加入到CE/EP混合物中,整体的模量有所降低,低温冻结后,树脂固化物间模量差加大,相对于其他试样,在一定的循环次数内树脂网络产生了过多的内应力积累,较容易达到树脂自身的载荷极限而导致出现裂纹、失效。进一步对比实验,制备纯CE试样和CE/EP/聚砜(用相同份数的聚砜(PSF)取代PKHH)试样,研究发现两者经过1次冷热循环后即出现裂纹,主要的原因可解释为,一方面,CE树脂自身的韧性差,固化网络无法有效分散应力载荷;另一方面,聚砜改性CE/EP固化物中形成分散相,分散相的存在能提高CE树脂韧性的同时[19],其与连续相基体的模量相差较大,冷热交替后产生大量内应力容易使两相界面破坏,从而导致试样出现裂纹。由此结果可知,CE树脂的增韧方式和改性组分的含量是影响其低温稳定性的关键因素。另外,由图8看出,CEPK10试样低温破坏的裂纹出现弯曲、分叉等现象,说明10%的PKHH有效地改善了CE树脂的低温稳定性。
综合考虑PKHH改性CE/EP树脂的固化特性、低温力学性能和热稳定性等,本研究认为5%~10%的PKHH改性CE/EP树脂体系具有较好的综合性能。
3 结论
(1)采用PKHH和EP树脂,通过熔融共混的方式共改性CE树脂,获得了兼具有良好热稳定性能、低温力学性能和优异的耐冷、热循环稳定性的改性氰酸酯树脂(CEPK)。与常规的热塑性树脂增韧CE树脂的机理不同,PKHH通过形成半互穿网络的方式改善了CE固化物的基本力学性能。
(2)PKHH能有效地提高CE树脂的耐低温性能,10%PKHH的加入可使CE树脂的–196 ℃冲击强度提高约40%,并且对CE/EP树脂的模量影响较小,10%PKHH的加入可使CE树脂经过冷、热循环首次出现裂纹的次数提高近5倍。
[1] 王珂, 虞鑫海, 徐永芬. 耐高温环氧树脂胶粘剂的研究进展[J]. 粘接, 2013, 34(2): 63-65.
(WANG K, YU X H, XUN Y F. Research progress of epoxy adhesives with high-temperature resistance[J].Adhesion, 2013, 34(2): 63-65.)
[2]NAKANE H, NISHIJIMA S, FUJISHIRO H, et al.Thermal properties of epoxy resins at cryogenic temperatures[C]//AIP Conference Proceedings. New York: AIP,2002, 614(1): 211-220.
[3]NISHIJIMA S, OKADA Y, HONDA Y. Evaluation of epoxy resin by position annihilation for cryogenic use[M]//Advances in Cryogenic Engineering Materials.New York: Springer US, 1994: 1137-1144.
[4] 王晓洁, 梁国正, 张伟, 等. 氰酸酯树脂在航空航天领域应用研究进展[J]. 材料导报, 2005, 19(5): 70-72.
(WANG X J, LIANG G Z, ZHANG W, et al. Applications of cyanate ester resin to aircraft and aerospace[J].Materials Review, 2005, 19(5): 70-72.)
[5]KIM E P, GRAFA N A, ELY K W. Cyanate ester composites for oxygen containment: US 6334589[P]. 2002-1-1.
[6] 赵红振, 齐暑华, 周文英, 等. 氰酸酯树脂增韧改性研究[J]. 热固性树脂, 2006, 21(2): 49-53.
(ZHAO H Z, QI S H, ZHOU W Y, et al. Research in toughening modification of cyanate ester resin[J]. Thermosetting Resin, 2006, 21(2): 49-53.)
[7] 谭京生. 环氧树脂超低温胶粘剂[J]. 中国胶粘剂, 2006,15(1): 32-36.
(TAN J S. Cryogenic epoxy resin adhesion[J]. China Adhesion, 2006, 15(1): 32-36.)
[8]WANG G, WANG R, FU G, et al. Study on phenolphthalein poly (ether sulfone)-modified cyanate ester resin and epoxy resin blends[J]. Polymer Engineering and Science, 2015, 55(11): 2591-2602.
[9]UEKI T, NISHIJIMA S, IZUMI Y. Designing of epoxy resin systems for cryogenic use[J]. Cryogenics, 2005,45(2): 141-148.
[10]UEKI T, NOJIMA K, ASANO K, et al. Toughening of epoxy resin systems for cryogenic use[M]// Advances in Cryogenic Engineering Materials. New York: Springer US, 1998: 277-283.
[11]RYU S H, SIN J H, SHANMUGHARAJ A M. Study on the effect of hexamethylene diamine functionalized graphene oxide on the curing kinetics of epoxy nanocomposites[J]. European Polymer Journal, 2014, 52: 88-97.
[12]KOO K K, INOUE T, MIYASAKA K. Toughened plastics consisting of brittle particles and ductile matrix[J]. Polymer Engineering and Science, 1985,43(2): 119-122.
[13]HARISMENDY I, DEL RIO M, ECEIZA A, et al. Morphology and thermal behavior of dicyanate ester-polyetherimide semi-IPNS cured at different conditions[J].Journal of Applied Polymer Science, 2000, 76(7): 1037-1047.
[14]HARISMENDY I, DEL RIO M, MARIETA C, et al. Dicyanate ester-polyetherimidesemi-interpenetrating polymer networks. II. Effects of morphology on the fracture toughness and mechanical properties[J]. Journal of Ap-plied Polymer Science, 2001, 80(14): 2759-2767.
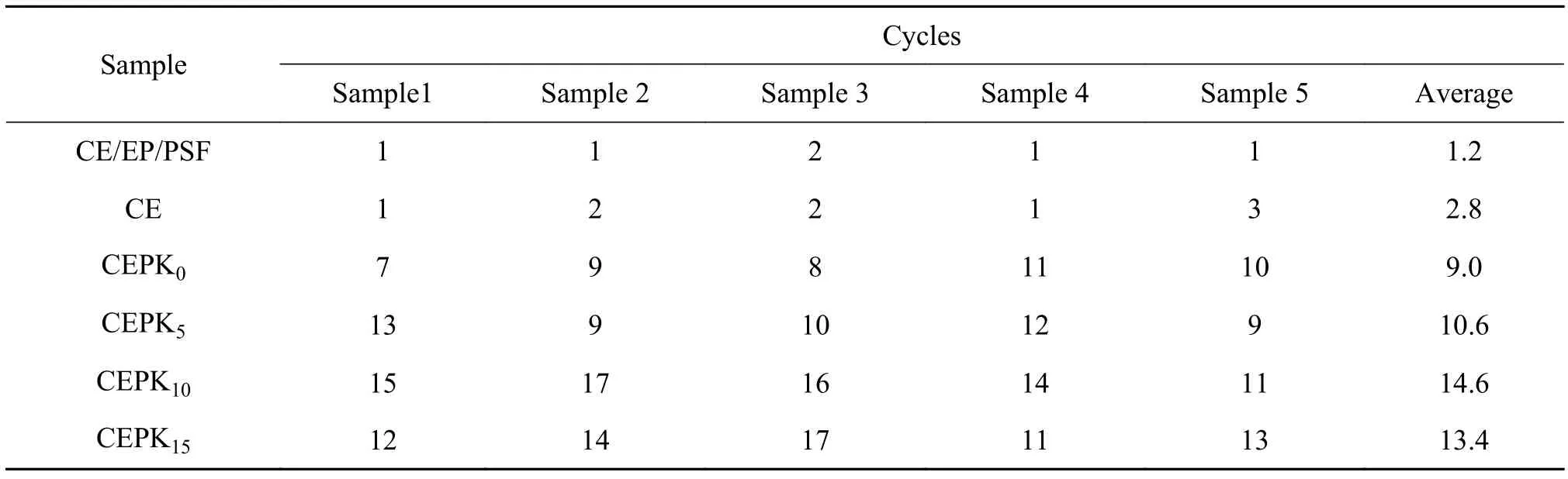
表5 不同树脂固化物经多次冷、热循环后表面首次出现裂纹情况(–196 ~200 ℃)Table 5 Crack states of different samples after several cycles (–196℃ to 200 ℃)
[15]李俊燕, 陈平, 马泽民, 等. 酚酞基聚醚酮/环氧树脂/氰酸酯半互穿聚合物的制备及性能[J]. 高分子材料科学与工程, 2009, 25(7): 141-143.
(LI J Y, CHEN P, MA Z M, et al. Preparation and characterization of EP/CE/PEK-C semi-IPN[J]. Polymer Materials Science & Engineering, 2009, 25(7): 141-143.)
[16]郭宝春, 邱清华, 贾德民. 互穿聚合网络(IPN)技术在高分子中的应用[J]. 功能材料, 2000, 31(3): 27-30.
(GUO B C, QIU Q H, JIA D M. Application of IPN technology in functional polymers[J]. Journal of Functional Materials, 2000, 31(3): 27-30.)
[17]过梅丽. 高聚物与复合材料的动态力学热分析[M]. 北京: 化学工业出版社, 2002: 37-38.
[18]MACKNIGHT W J, KARASZ F E, FRIED J R. Solid state transition behavior of blends[J]. Polymer Blends,1978, 1: 185-242.
[19]MONDRAGON I, SOLAR L, NOHALES A, et al. Properties and structure of cyanate ester/polysulfone/ organoclay nanocomposites[J]. Polymer, 2006, 47(10): 3401-3409.
