氧化钼/聚吡咯复合膜的制备及变色性能研究*
2018-04-10张文治王素敏
张 阁,张文治,王素敏
(西安工业大学 陕西省光电功能材料与器件重点实验室/材料与化工学院,西安 710021)
电致变色材料被认为是目前最有应用前景的智能材料之一.电致变色是指材料的光学属性(反射率、透过率和吸收率等)在外加电场的作用下发生稳定、可逆的颜色变化的现象,在外观上表现为颜色和透明度的可逆变化.利用电致变色薄膜在电场下对光线的调制作用,与平板玻璃制成建筑物的智能窗可实现对室内温度的调节,以节约能量,减少污染[1].
变色层是变色器件的核心层.在无机变色层中研究较多的是MoO3,与其他材料相比,三氧化钼(MoO3)薄膜具有良好的光致变色和电致变色(变色响应时间较短[2])性能,得到了广泛应用.人们采用多种方法合成不同形貌的三氧化钼,常用的方法有化学气相沉积法、溶胶-凝胶法和水热法等.文献[3]利用热丝化学气相沉积法在氩气氛围中合成了一维MoO11纳米棒,并通过调节沉积温度实现了MoO11纳米棒禁带宽度的调控.此方法可得到高纯度和高结晶性的氧化钼,但合成过程要求严格,通常需在高温高压和特殊气氛下进行,对环境要求较高,合成过程能耗较大.文献[4]将MoO3粉末与氨水/双氧水混合溶液作为溶胶-凝胶法的前驱体,并在室温下冷却、陈化获得MoOx凝胶,利用旋涂和紫外线(Ultraviolet,UV)辐照在氧化铟锡(Indium Tin Oxide,ITO)衬底上制备了MoOx纳米薄膜.溶胶-凝胶法与其他氧化钼纳米材料的合成方法相比,具有反应温度低,易掺杂,产物均匀和易制备新结构等优点.但溶胶-凝胶法合成的原料大多为有机物对健康有害,且合成过程时间较长,效率相对较低.文献[5]将钼箔溶解在双氧水溶剂中获得钼酸盐溶剂,以钼酸盐溶剂作电解质进行电化学沉积,该方法获得的MoO3薄膜的尺寸为40~100 nm,且薄膜致密而平滑.电化学沉积适用于各种形状的基体材料,该方法工艺简单,操作容易,环境安全,非常适用于工业化量产.但对于基体表面氧化钼晶核的生成和长大速度难以控制,因而获得的氧化钼薄膜多为多晶态或非晶态,材料质量不够高.水热合成法相比其他方法操作简单,合成方便,成为研究重点.聚吡咯(Polypyrrole,PPy)作为一种导电聚合物,因其环境稳定性好、易合成、无毒、氧化还原可逆、变色速度快和着色效率高等[6-8],在电致变色领域具有较好的应用前景[9-10].但聚吡咯电致变色器件存在循环寿命低和结构致密等缺点,这限制了其在光电器件中的应用.无机变色材料组装的变色器件循环寿命长,但颜色单一,响应速度慢.基于两者存在的问题,研究者们提出了互补型电致变色器件,其相对于单一电致变色器件具有循环寿命长,光调制范围大,能最大限度的调节透过率等优点而被广泛应用.为研究复合变色器件的性能,本文选用水热合成法成功制得三氧化钼纳米棒,通过恒压法在ITO玻璃上制得聚吡咯薄膜.随后将氧化钼薄膜作为阴极变色层与聚吡咯阳极变色层复合组装成互补型电致变色器件.
1 实验部分
1.1 三氧化钼纳米晶的制备
称取0.588 5 g七钼酸铵粉末溶于10 mL去离子水中,搅拌15 min后,在七钼酸铵水溶液中缓慢加入2.422 mL的浓硝酸(HNO3);搅拌均匀后,将其混合物移至100 mL不锈钢高压反应釜中,90 ℃下反应3 h,待体系冷却至室温,用去离子水和无水乙醇多次洗涤离心,收集沉淀物;将沉淀物在电热鼓风干燥箱中,60 ℃下干燥5 h,然后将其移至真空干燥箱中,60 ℃下干燥12 h,即得样品.
1.2 三氧化钼浆料及薄膜的制备
在样品瓶中,称取0.05 g乙基纤维素,然后加入6 mL无水乙醇,待其溶解后,加入0.1 g氧化钼和0.44 mL松油醇,充分搅拌直至无水乙醇挥发完全制得浆料.将制好的浆料涂覆在ITO导电玻璃表面,将其放入管式高温烧结炉中加热制得薄膜.管式炉升温程序设置为:5 ℃·min-1速率升温至325 ℃,在此温度下恒温烧结5 min,以2 ℃·min-1速率升温至375 ℃,此温度下恒温烧结5 min,以2 ℃·min-1速率升温至450 ℃,此温度下恒温烧结15 min,以2 ℃·min-1速率升温至480 ℃,此温度下恒温烧结15 min后,自然降至室温.
1.3 聚吡咯薄膜的制备
在烧杯中加入50 mL蒸馏水,然后加入0.86 g对甲苯磺酸,采用320 r·min-1的搅拌速率,搅拌混合10 min;之后,加入0.335 g吡咯单体,转速调整为500 r·min-1,搅拌混合5 min.将ITO玻璃的导电面,与Pt电极相对放置,并将电极浸没在吡咯溶液中;采用恒电压法聚合制得聚吡咯薄膜.将其放入干燥箱中,60 ℃下干燥3 h.
1.4 三氧化钼/聚吡咯复合电致变色器件的组装
在小烧杯中加入2.8 g乙腈和0.05 g无水高氯酸锂,待充分溶解后,加入1.0 g PMMA和0.8 g PC,超声1 h制得凝胶电解质.在表面涂有三氧化钼薄膜的ITO玻璃上,滴加凝胶电解质溶液.然后将聚吡咯薄膜作为对电极,与三氧化钼薄膜电极组装成三明治结构,得到三氧化钼/聚吡咯复合电致变色器件.
1.5 测试方法
采用日本津岛XRD-6000 型X射线衍射仪(X-Ray Diffraction,XRD)测试晶体的相结构,测试条件为Cu靶Kα(λ=1 154 nm),扫描范围(2θ)为5°~60°;用美国Thermo Nicolet型傅里叶变换红外光谱仪(Fourier Transform Infrared Spectrometer,FT-IR)对样品进行测试,根据化学键或官能团分析样品的组成与结构,测试范围4 000~400 cm-1;用瑞士万通的PGSTAT 302 N型电化学工作站进行循环伏安测试,其中铂电极作为对电极,Ag/AgCl电极为参比电极,被测薄膜为工作电极;紫外-可见光谱仪(Shimadzu 2550)与瑞士万通的PGSTAT 302 N型电化学工作站联用来测试器件的电致变色性能,通过电化学工作站提供电压,结合紫外-可见光谱仪测试器件在不同电位下的吸收光谱及变色动力学曲线.
2 结果与分析
2.1 MoO3的X射线衍射分析
图1为硝酸量2.422 mL,不同反应时间下制得的MoO3纳米晶的XRD谱图,产物与标准谱卡(JCPDS卡21-0569)的衍射峰位基本一致,所以确定产物即为MoO3,且各衍射峰均归属为六方相MoO3(h-MoO3);没有其他杂峰出现,表明产物纯度较高.此外,随着反应时间的延长,衍射峰的位置基本没有变化,但衍射峰的强度随反应时间的延长而增强,特别是(210)、(300)和(008)晶面对应的衍射峰强度较高,可能是由于随着反应时间的延长,晶体生长择优取向,沿一维方向生长,晶体更加完善.而且在16.7°即(110)晶面,可以看到随反应时间的延长,衍射峰的强度逐渐增强,进一步证实晶体生长更趋完善.
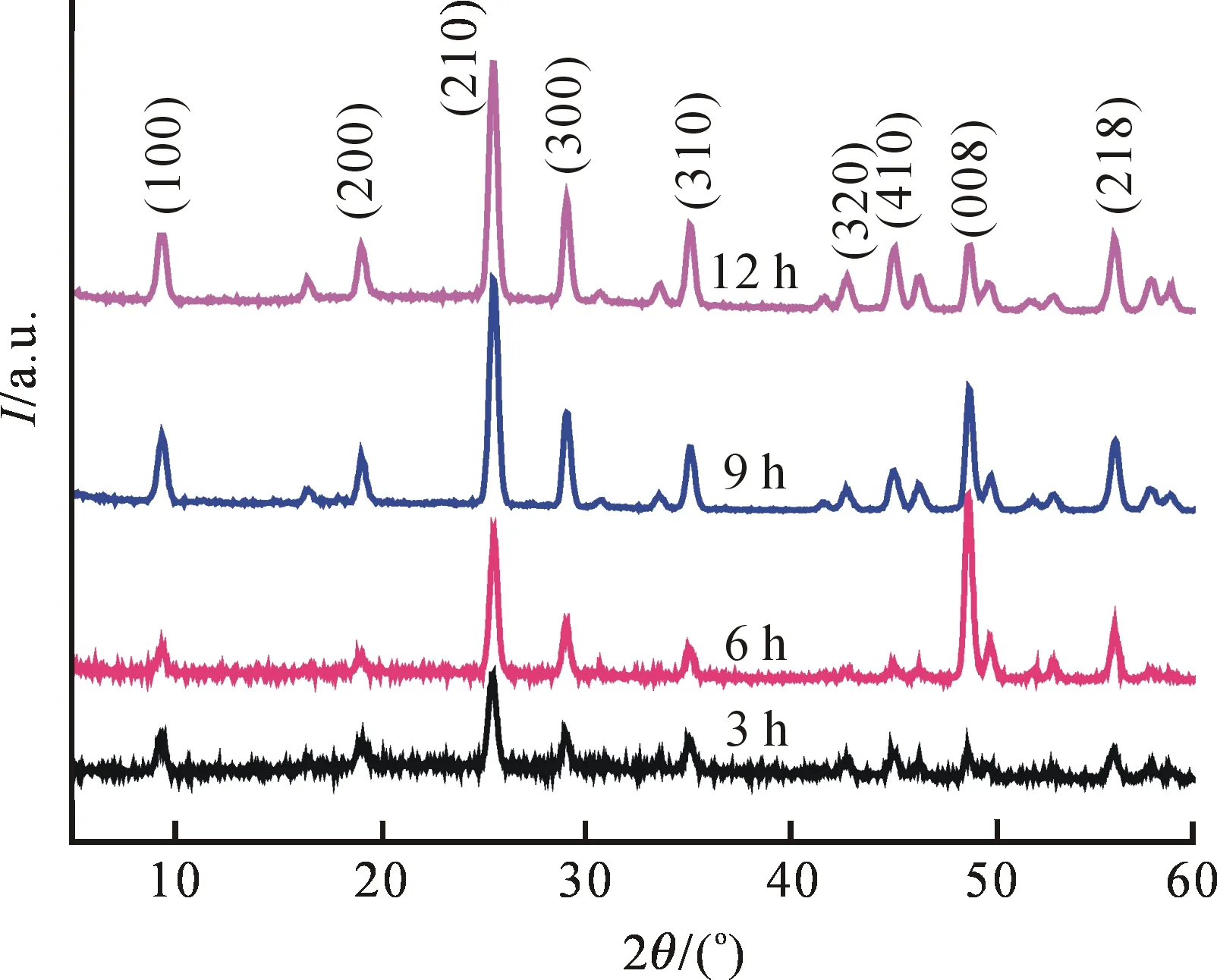
图1 MoO3纳米晶的XRD谱图
2.2 MoO3的红外谱图分析
图2为不同水热反应时间下制得的MoO3的红外谱图.

图2 MoO3纳米晶的红外谱图
从图2中可看到,在974 cm-1和911 cm-1处的吸收峰归属于Mo=O双键的伸缩振动峰,612 cm-1处的吸收峰为Mo-O的伸缩振动峰,1 383 cm-1处的吸收峰为Mo-OH的伸缩振动峰[11].3 410 cm-1和1 615 cm-1处出现的吸收峰分别归属于水分子中-OH的伸缩振动峰和弯曲振动峰.综上可知,文中成功合成了MoO3纳米晶.从图2还可以看出,随着反应时间的延长,位于1 383 cm-1处的吸收峰向长波数方向移动;当反应时间超过6 h时,Mo-OH的伸缩振动峰移动到1 403 cm-1,而且911 cm-1处的Mo=O双键伸缩振动峰移动向长波数移动至913 cm-1(反应时间为12 h),吸收峰的移动可能是由反应时间的延长使得氧化钼晶体逐渐生长趋于完善所导致的.
2.3 MoO3的扫描电子显微镜分析
当反应温度为90 ℃,硝酸加入量为2.422 mL时,本文研究了不同水热反应时间对MoO3纳米晶形貌的影响.当反应时间为9 h时,从图3(a)的低倍照片中可以看到,生成的MoO3六角微米棒较少.从高倍照片中可以看到,形成的六角微米棒形状比较规整,且表面比较光滑,其直径为3 μm左右,长度为20 μm左右,长径比较大.延长反应时间至12 h时,从图3(b)的低倍照片中可以看到,一部分小碎棒长成较完整的六角微米棒,六角棒的数量增多.从高倍照片中可以看到,六角微米棒的直径和尺寸变大,说明反应时间对MoO3的形貌有一定的影响.随着反应时间的延长,晶体形成更加完善且数量增多.
2.4 MoO3的循环伏安曲线分析
采用电化学工作站对MoO3薄膜的电化学性能进行表征.测试时采用标准三电极体系,以氧化钼薄膜/ITO玻璃为工作电极,饱和甘汞电极作参比电极,铂片为对电极,电解液为0.1 mol·L-1高氯酸锂的乙腈溶液.图4为水热反应时间12 h下制得的MoO3薄膜的循环伏安曲线.

图3 水热反应时间为(a) 9 h和(b) 12 h下制得的MoO3的SEM照片
从图4中可以看出,扫描速率为100 mV·s-1时,在-0.06 V左右出现氧化峰,-0.26 V左右出现还原峰,但峰形不明显,且CV的闭合曲线面积较小,表明其导电性较差,这就使得其电化学性能受到限制.因此本文采用聚吡咯与氧化钼复合,形成互补型电致变色器件有可能提高其应用价值.

图4 MoO3薄膜的循环伏安曲线
2.5 MoO3器件的紫外-可见光谱分析
从图5中可以看出,氧化钼器件的紫外-可见光谱中出现两个吸收峰,分别位于373 nm和436 nm,其中436 nm处的吸收峰较明显.当外加电压从-2.5 V升高到+2.5 V时,该处吸收峰的强度从0.346增大到0.360,光学对比度变化不大.表明MoO3器件具有一定的电致变色性能.当外加电压为+2.5 V时,器件的颜色较深;当电压较低时,其吸光度基本不变(紫外-可见吸收曲线重合),这是由于低电压不足以驱动MoO3器件中变色薄膜发生氧化还原反应,故变色效果不明显.
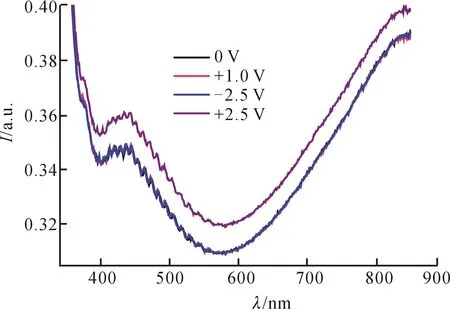
图5 MoO3器件在不同电压下的紫外-可见光谱
2.6 聚吡咯的红外光谱分析
在图6中,1 546 cm-1处的吸收峰为C=C和C-C组合的伸缩振动峰,1 469 cm-1处的特征吸收峰归属于C-N的伸缩振动峰,1 302 cm-1处的特征吸收峰为N-H键的弯曲振动峰,911 cm-1处的吸收峰归属于吡咯环C-H键的面外弯曲振动峰.以上分析表明,文中成功合成了聚吡咯.

图6 聚吡咯的红外光谱图
2.7 MoO3/聚吡咯复合器件的电致变色性能
图7为所制得的氧化钼/聚吡咯复合器件的照片.图8为氧化钼/聚吡咯复合器件在不同电压下的紫外-可见光谱,从图8中可看到,当外加电压从-1.5 V升高到+1.5 V时,481 nm处吸收峰的光学对比度变化最大,其强度从0.62减小到0.55,表明MoO3/聚吡咯复合器件具有较好的电致变色性能.当外加电压为+1.5 V时,器件的颜色变深,电致变色现象明显.

图7 氧化钼/聚吡咯复合器件的照片
除了对比度外,响应时间也是判断材料电致变色性能的重要参数.图9为在电压范围-1.5~+1.5 V的方波电压下对MoO3/聚吡咯复合器件进行动力学测试得到的曲线.其中,方波电压的循环周期为100 s,测试得到的为器件在481 nm处吸光度随时间的变化曲线.持续9圈后,动力学曲线无明显衰减,但曲线整体向下偏移,这主要是由于复合器件中的氧化钼存在电致变色“滞后现象”,器件中活性层发生氧化还原反应变色的速率滞后于电压的变化.此外,可以看到器件变色的重现性较好.根据动力学曲线,可以得到复合器件发生变色时的着色时间为35 s,褪色时间为22 s.其中,着色态颜色为浅蓝色,褪色态颜色为淡黄色.
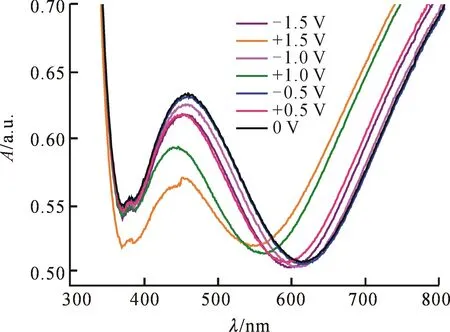
图8 氧化钼/聚吡咯复合器件在不同电压下的紫外-可见光谱
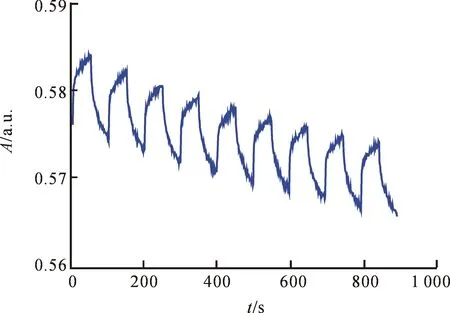
图9 氧化钼/聚吡咯复合器件的变色动力学曲线
3 结 论
1) 采用水热法成功制得了h-MoO3,且氧化钼的晶型不随水热反应时间而变化;SEM分析表明,合成的MoO3呈现六方棒状.MoO3薄膜的CV曲线表明,其氧化峰和还原峰分别位于-0.06 V和-0.26 V;制得的氧化钼器件在外加电压为+2.5 V时,颜色发生变化.
2) 氧化钼/聚吡咯复合器件的UV-vis光谱测试表明,当外加电压从-1.5 V升高到+1.5 V时,481 nm处吸收峰的强度从0.62减小到0.55,光学对比度变化较大.当外加电压为+1.5 V时,器件的颜色变深.其着色时间为35 s,褪色时间为22 s;着色态颜色为浅蓝色,褪色态颜色为淡黄色.
参考文献:
[1]郭荣辉,杨晓青,秦文峰.电致变色三氧化钼薄膜研究进展[J].中国钼业,2005,29(5):39.
GUO Ronghui,YANG Xiaoqing,QIN Wenfeng.Research Progress of electrochromic molybdenum trioxide thin films[J].China Molybdenum Industry,2005,29(5):39.(in Chinese)
[2]CRUZ T G S,GORENSTEIN A,LANDERS R,et al.Electrochromism in MoOxFilms Characterized by X-ray Electron Spectroscopy[J].Journal of Electron Spectroscopy & Related Phenomena,1999,101(98):397.
[3]PHAM D V,PATIL R A,LIN J H,et al.Doping-free Bandgap Tuning in One-dimensional Magnéli-phase Nanorods of Mo4O11[J].Nanoscale,2016,8(10):5559.
[4]FU Q,CHEN J S,SHI C S,et al.Room-Temperature Sol-Gel Derived Molybdenum Oxide Thin Films for Efficient and Stable Solution-Processed Organic Light-Emitting Diodes[J].ACS Applied Materials & Interfaces,2013,5(13):6024.
[5]TODD M M,STEVENSON K J,HUPP J T,et al.Electrochemical Preparation of Molybdenum Trioxide Thin Films:Effect of Sintering on Electrochromic and Electroinsertion Properties[J].Langmuir,2003,19(10):4316.
[6]CARRASCO P M,GRANDE H J,CORTAZAR M,et al.Structure-Conductivity Relationships in Chemical Polypyrroles of Low,Medium and High Conductivity[J].Synthetic Metals,2006,156(5):420.
[7]WHANG Y E,HAN J H,MOYOBE T,et al.Polypyrroles Prepared by Chemical Oxidative Polymerization at Different Oxidation Potentials[J].Synthetic Metals,1991,45(2):151.
[8]李倩倩,黄健涵,刘素琴,等.聚吡咯的化学氧化合成及其对金属镁的防腐蚀性能研究[J].功能材料,2008,39(3):526.
LI Qianqian,HUANG Jianhan,LIU Suqin,et al,Chemical Oxidative Polymerization of PPy and Its Corrosion Resistance on the Surface of Metals[J].Journal of Functional Materials,2008,39(3):526.
(in Chinese)
[9]MALINAUSKAS A.Chemical Deposition of Conducting Polymers[J].Polymer,2001,42(9):3957.
[10]MAHESWARI N,MURALIDHARAN G.Controlled Synthesis of Nanostructured Molybdenum Oxide Electrodes for High Performance Supercapacitor Devices[J].Applied Surface Science,2017,416:461.
[11]CHITHAMBARAJ A,YOGAMALAR N R,BOSE A C.Hydrothermally Synthesized h-MoO3 and#-MoO3 Nanocrystals:New Findings on Crystal-Structure-Dependent Charge Transport[J].Crystal Growth & Design,2016,16(4):1984.
